57 Search Results for "animal models"
Healing Switch Links Acute Kidney Injury to Fibrosis, Suggesting Way to Protect Kidney Function
Posted on by Dr. Monica M. Bertagnolli
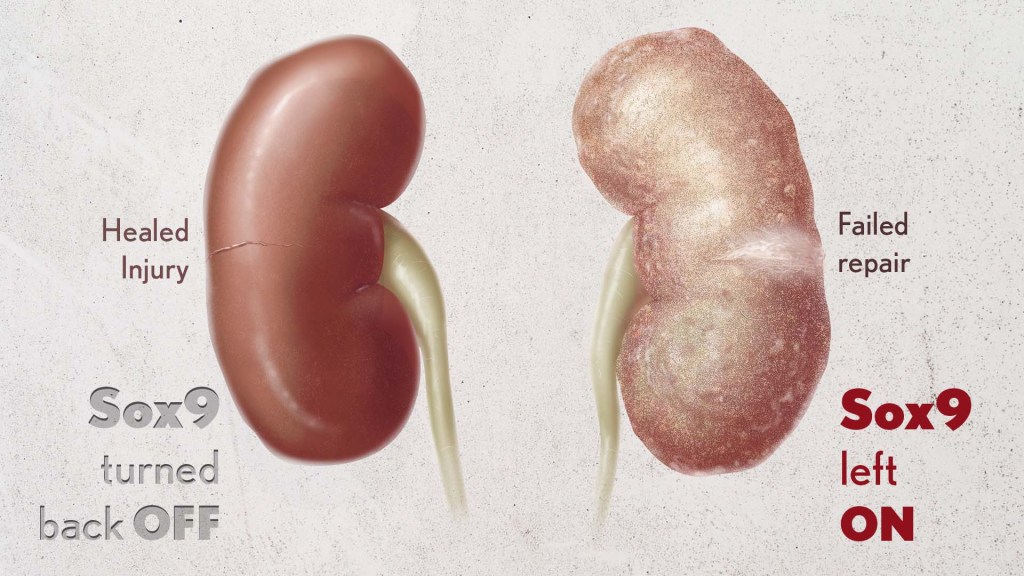
Healthy kidneys—part of the urinary tract—remove waste and help balance chemicals and fluids in the body. However, our kidneys have a limited ability to regenerate healthy tissue after sustaining injuries from conditions such as diabetes or high blood pressure. Injured kidneys are often left with a mix of healthy and scarred tissue, or fibrosis, which over time can compromise their function and lead to chronic kidney disease or complete kidney failure. More than one in seven adults in the U.S. are estimated to have chronic kidney disease, according to the Centers for Disease Control and Prevention, most without knowing it.
Now, a team of researchers led by Sanjeev Kumar at Cedars-Sinai Medical Center, Los Angeles, has identified a key molecular “switch” that determines whether injured kidney tissue will heal or develop those damaging scars.1 Their findings, reported in the journal Science, could lead to new and less invasive ways to detect fibrosis in the kidneys. The research could also point toward a targeted therapeutic approach that might prevent or reverse scarring to protect kidney function.
In earlier studies, the research team found that a protein called Sox9 plays an important role in switching on the repair response in kidneys after acute injury.2 In some cases, the researchers noticed that Sox9 remained active for a prolonged period of a month or more. They suspected this might be a sign of unresolved injury and repair.
By conducting studies using animal models of kidney damage, the researchers found that cells that turned Sox9 on and then back off healed without fibrosis. However, cells that failed to regenerate healthy kidney cells kept Sox9 on indefinitely, which in turn led to the production of fibrosis and scarring.
According to Kumar, Sox9 appears to act like a sensor, switching on after injury. Once restored to health, Sox9 switches back off. When healing doesn’t proceed optimally, Sox9 stays on, leading to scarring. Importantly, the researchers also found they could encourage kidneys to recover by forcing Sox9 to turn off a week after an injury, suggesting it may be a promising drug target.
The researchers also looked for evidence of this process in human patients who have received kidney transplants. They could see that, when transplanted kidneys took longer to start working, Sox9 was switched on. Those whose kidneys continued to produce Sox9 also had lower kidney function and more scarring compared to those who didn’t.
The findings suggest that the dynamics observed in animal studies may be clinically relevant in people, and that treatments targeting Sox9 might promote kidneys to heal instead of scarring. The researchers say they hope that similar studies in the future will lead to greater understanding of healing and fibrosis in other organs—including the heart, lungs, and liver—with potentially important clinical implications.
References:
[1] Aggarwal S, et al. SOX9 switch links regeneration to fibrosis at the single-cell level in mammalian kidneys. Science. DOI: 10.1126/science.add6371 (2024).
[2] Kumar S, et al. Sox9 Activation Highlights a Cellular Pathway of Renal Repair in the Acutely Injured Mammalian Kidney. Cell Reports. DOI: 10.1016/j.celrep.2015.07.034 (2015).
NIH Support: National Institute of Diabetes and Digestive and Kidney Diseases
Pain Circuit Discovery in the Brain Suggests Promising Alternative to Opioid Painkillers
Posted on by Lawrence Tabak, D.D.S., Ph.D.
Chronic pain is an often-debilitating health condition and serious public health concern, affecting more than 50 million Americans.1 The opioid and overdose crisis, which stems from inadequate pain treatment, continues to have a devastating impact on families and communities across the country. To combat both challenges, we urgently need new ways to treat acute and chronic pain effectively without the many downsides of opioids.
While there are already multiple classes of non-opioid pain medications and other approaches to manage pain, unfortunately none have proved as effective as opioids when it comes to pain relief. So, I’m encouraged to see that an NIH-funded team now has preclinical evidence of a promising alternative target for pain-relieving medicines in the brain.2
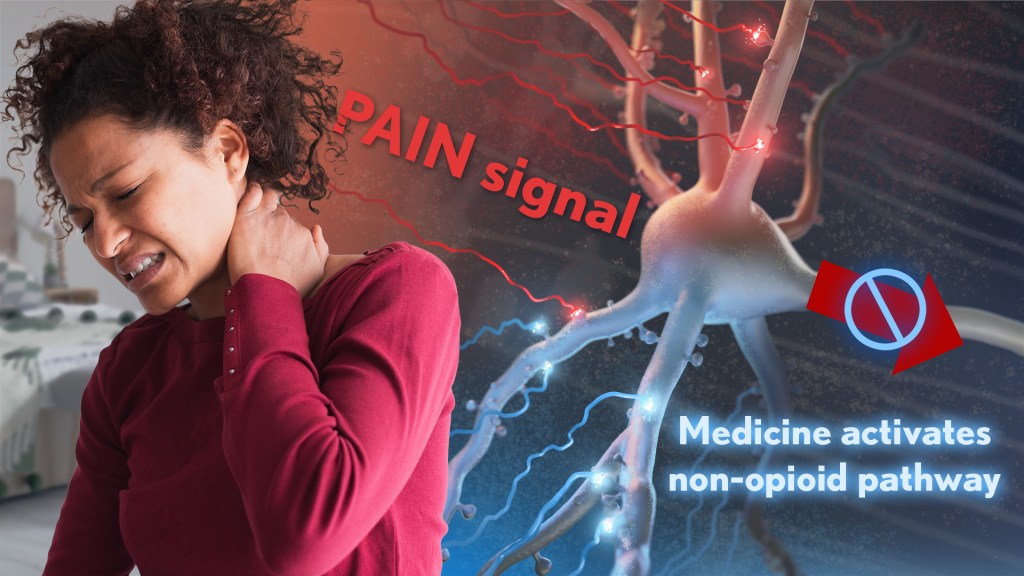
Rather than activating opioid receptors, the new approach targets receptors for a nerve messenger known as acetylcholine in a portion of the brain involved in pain control. Based on findings from animal models, it appears that treatments targeting acetylcholine could offer pain relief even in people who have reduced responsiveness to opioids. Their findings suggest that the treatment approach has the potential to remain effective in combatting pain long-term and with limited risk for withdrawal symptoms or addiction.
The researchers, led by Daniel McGehee, University of Chicago, focused their attention on non-opioid pathways in the ventrolateral periaqueductal gray (vlPAG), a brain area involved in pain control. They had previously shown that activating acetylcholine receptors, which are part of the vlPAG’s underlying circuitry, could relieve pain.3 However, they found that when the body is experiencing pain, it unexpectedly suppresses acetylcholine rather than releasing more.
To understand how and why this is happening, McGehee and Shivang Sullere, now a postdoctoral fellow at Harvard Medical School, conducted studies in mice to understand how acetylcholine is released under various pain states. They found that mice treated with a drug that stimulates an acetylcholine receptor known as alpha-7 (⍺7) initially led to more activity in the nervous system. But this activity quickly gave way to a prolonged inactive or quiet state that delivered pain relief. Interestingly, this unexpected inhibitory effect lasted for several hours.
Additional studies in mice that had developed a tolerance to opioids showed the same long-lasting pain relief. This encouraging finding was expected since opioids use a pathway separate from acetylcholine. In more good news, the animals didn’t show any signs of dependence or addiction either. For instance, in the absence of pain, they didn’t prefer spending time in environments where they’d receive the ⍺7-targeted drug.
Imaging studies measuring brain activity revealed greater activity in cells expressing ⍺7 with higher levels of pain. When that activity was blocked, pain levels dropped. The finding suggests to the researchers it may be possible to monitor pain levels through brain imaging. It’s also possible the acetylcholine circuitry in the brain may play a role in the process whereby acute or temporary pain becomes chronic.
Finding treatments to modify acetylcholine levels or target acetylcholine receptors may therefore offer a means to treat pain and prevent it from becoming chronic. Encouragingly, drugs acting on these receptors already have been tested for use in people for treating other health conditions. It will now be important to learn whether these existing therapeutics or others like them may act as highly effective, non-addictive painkillers, with important implications for alleviating chronic pain.
References:
[1] NIH HEAL Initiative Research Plan. NIH HEAL Initiative.
[2] Sullere S et al. A cholinergic circuit that relieves pain despite opioid tolerance. Neuron. DOI: 10.1016/j.neuron.2023.08.017 (2023).
[3] Umana IC et al. Nicotinic modulation of descending pain control circuitry. Pain. PMID: 28817416; PMCID: PMC5873975 (2017).
Links:
The Helping to End Addiction Long-term® (HEAL) Initiative (NIH)
Pain (National Institute of Neurological Disorders and Stroke/NIH)
Opioids (National Institute on Drug Abuse/NIH)
Daniel McGehee (University of Chicago, Illinois)
NIH Support: National Institute of Neurological Disorders and Stroke, National Institute on Drug Abuse
Unraveling the Role of the Skin Microbiome in Health and Disease
Posted on by Lindsey A. Criswell, M.D., M.P.H., D.Sc., National Institute of Arthritis and Musculoskeletal and Skin Diseases

Human skin is home to diverse ecosystems including bacteria, viruses, and fungi. These microbial communities comprise hundreds of species and are collectively known as the skin microbiome. The skin microbiome is thought to play a vital role in fending off disease-causing microorganisms (pathogens), boosting barrier protection, and aiding immune defenses.
Maintaining a balanced skin microbiome involves a complex and dynamic interplay among microorganisms, immune cells, skin cells, and other factors. In general, bacteria far outnumber viral, fungal, or other microbial species on the skin. Bacterial communities, which are strongly influenced by conditions such as skin moisture, temperature, and pH, vary widely across the body. For example, facial cheek skin hosts mostly Cutibacterium along with a bit of the skin fungus Malassezia. The heel is colonized by different types of bacteria including Staphylococcus and Corynebacteria.
In some diseases, such as acne and eczema, the skin microbiome is altered. Typically, this means an increase in pathogenic microorganisms and a decrease in beneficial ones. An altered skin microbiome can also be associated with inflammation, severe disease symptoms, and changes in the human immune system.
Heidi H. Kong is working to understand the role of the skin microbiome in health and disease. She is a senior investigator in the Intramural Research Program at NIH’s National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) and an adjunct investigator at NIH’s National Cancer Institute (NCI).
More than a decade ago, Kong and Julie A. Segre, an intramural researcher at NIH’s National Human Genome Research Institute, analyzed the microbial makeup of healthy individuals. Kong swabbed the skin of these healthy volunteers in 20 different sites, from the forehead to the toenail. The study revealed that the surface of the human body provides various environmental niches, depending on whether the skin is moist, dry, or sebaceous (oily). Different bacterial species predominate in each niche. Kong and Segre were particularly interested in body areas that have predilections for disease. For example, psoriasis is often found on the outside of elbows and knees, and the back of the scalp.
Earlier this year, Kong and Segre published another broad analysis of the human skin microbiome [1] in collaboration with scientists at the European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), United Kingdom. This new catalog, called the Skin Microbial Genome Collection, is thought to identify about 85 percent of the microorganisms present on healthy skin from 19 body sites. It documents more than 600 bacterial species—including 174 that were discovered during the study—as well as more than 6,900 viruses and some fungi, including three newly discovered species.
Kong’s work has provided compelling evidence that the human immune system plays a role in shaping the skin microbiome. In 2018, she, Segre, and colleagues from the intramural programs of NCI and NIH’s National Institute of Allergy and Infectious Diseases analyzed skin from eight different sites on 27 people with a rare primary immunodeficiency disease known as DOCK8 deficiency [2].
People with the condition have recurrent infections in the skin, sinuses, and airways, and are susceptible to different cancers. Kong and colleagues found that the skin of people with DOCK8 deficiency contains significantly more DNA viruses (90 percent of the skin microbiome on average) than people without the condition (6 or 7 percent of the skin microbiome).
Other researchers are hoping to leverage features of the microbiome to develop targeted therapies for skin diseases. Richard L. Gallo, a NIAMS grantee at the University of California, San Diego, is currently focused on acne and eczema (also called atopic dermatitis). Acne is associated with certain strains of Cutibacterium acnes (C. acnes, formerly called Propionibacterium acnes or P. acnes). Eczema is often associated with Staphylococcus aureus (S. aureus).
Severe cases of acne and eczema are commonly treated with broad-spectrum antibiotics, which wipe out most of the bacteria, including beneficial species. The goal of microbiome-targeted therapy is to kill only the disease-associated bacteria and avoid increasing the risk that some strains will develop antibiotic resistance.
In 2020, Gallo and colleagues identified a strain of Staphylococcus capitis from healthy human skin (S. capitis E12) that selectively inhibits the growth of C. acnes without negatively impacting other bacteria or human skin cells [3]. S. capitis E12 produces four different toxins that act together to target C. acnes. The research team created an extract of the four toxins and tested it using animal models. In most cases, the extract was more potent at killing C. acnes—including acne-associated strains—than several commonly prescribed antibiotics (erythromycin, tetracycline, and clindamycin). And, unlike antibiotics, the extract does not appear to promote drug-resistance, at least for the 20 generations observed by the researchers.
Eczema is a chronic, relapsing disease characterized by skin that is dry, itchy, inflamed, and prone to infection, including by pathogens such as S. aureus and herpes virus. Although the cause of eczema is unknown, the condition is associated with human genetic mutations, disruption of the skin’s barrier, inflammation-triggering allergens, and imbalances in the skin microbiome.
In 2017, Gallo’s research team discovered that, in healthy human skin, certain strains of Staphylococcus hominis and Staphylococcus epidermis produce potent antimicrobial molecules known as lantibiotics [4]. These beneficial strains are far less common on the skin of people with eczema. The lantibiotics work synergistically with LL-37, an antimicrobial molecule produced by the human immune system, to selectively kill S. aureus, including methicillin-resistant strains (MRSA).
Gallo and his colleagues then examined the safety and therapeutic potential of these beneficial strains isolated from the human skin microbiome. In animal tests, strains of S. hominis and S. epidermis that produce lantibiotics killed S. aureus and blocked production of its toxin.
Gallo’s group has now expanded their work to early studies in humans. In 2021, two independent phase 1 clinical trials [5,6] conducted by Gallo and his colleagues investigated the effects of these strains on people with eczema. These double-blind, placebo-controlled trials involved one-week of topical application of beneficial bacteria to the forearm of adults with S. aureus-positive eczema. The results demonstrated that the treatment was safe, showed a significant decrease in S. aureus, and improved eczema symptoms in most patients. This is encouraging news for those hoping to develop microbiome-targeted therapy for inflammatory skin diseases.
As research on the skin microbiome advances on different fronts, it will provide deeper insight into the multi-faceted microbial communities that are so critical to health and disease. One day, we may even be able to harness the microbiome as a source of therapeutics to alleviate inflammation, promote wound healing, or suppress certain skin cancers.
References:
[1] Integrating cultivation and metagenomics for a multi-kingdom view of skin microbiome diversity and functions. Saheb Kashaf S, Proctor DM, Deming C, Saary P, Hölzer M; NISC Comparative Sequencing Program, Taylor ME, Kong HH, Segre JA, Almeida A, Finn RD. Nat Microbiol. 2022 Jan;7(1):169-179.
[2] Expanded skin virome in DOCK8-deficient patients. Tirosh O, Conlan S, Deming C, Lee-Lin SQ, Huang X; NISC Comparative Sequencing Program, Su HC, Freeman AF, Segre JA, Kong HH. Nat Med. 2018 Dec;24(12):1815-1821.
[3] Identification of a human skin commensal bacterium that selectively kills Cutibacterium acnes. O’Neill AM, Nakatsuji T, Hayachi A, Williams MR, Mills RH, Gonzalez DJ, Gallo RL. J Invest Dermatol. 2020 Aug;140(8):1619-1628.e2.
[4] Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, Shafiq F, Kotol PF, Bouslimani A, Melnik AV, Latif H, Kim JN, Lockhart A, Artis K, David G, Taylor P, Streib J, Dorrestein PC, Grier A, Gill SR, Zengler K, Hata TR, Leung DY, Gallo RL. Sci Transl Med. 2017 Feb 22;9(378):eaah4680.
[5] Development of a human skin commensal microbe for bacteriotherapy of atopic dermatitis and use in a phase 1 randomized clinical trial. Nakatsuji T, Hata TR, Tong Y, Cheng JY, Shafiq F, Butcher AM, Salem SS, Brinton SL, Rudman Spergel AK, Johnson K, Jepson B, Calatroni A, David G, Ramirez-Gama M, Taylor P, Leung DYM, Gallo RL. Nat Med. 2021 Apr;27(4):700-709.
[6] Use of autologous bacteriotherapy to treat Staphylococcus aureus in patients with atopic dermatitis: A randomized double-blind clinical trial. Nakatsuji T, Gallo RL, Shafiq F, Tong Y, Chun K, Butcher AM, Cheng JY, Hata TR. JAMA Dermatol. 2021 Jun 16;157(8):978-82.
Links:
Acne (National Institute of Arthritis and Musculoskeletal and Skin Diseases/NIH)
Atopic Dermatitis (NIAMS)
Cutaneous Microbiome and Inflammation Laboratory, Heidi Kong (NIAMS)
Julie Segre (National Human Genome Research Institute/NIH)
Gallo Lab (University of California, San Diego)
[Note: Acting NIH Director Lawrence Tabak has asked the heads of NIH’s Institutes and Centers (ICs) to contribute occasional guest posts to the blog to highlight some of the cool science that they support and conduct. This is the fifth in the series of NIH IC guest posts that will run until a new permanent NIH director is in place.]
Finding the ‘Tipping Point’ to Permanent Kidney Damage
Posted on by Lawrence Tabak, D.D.S., Ph.D.
Healthy human kidneys filter more than 30 gallons of blood each day on average, efficiently removing extra fluid and harmful toxins from the body. If injured, the kidneys have a remarkable capacity for repair. And, yet, in more than one in seven U.S. adults, including disproportionately people with diabetes and hypertension, the daily wear and tear on these vital organs has passed a “tipping point” toward irreparable damage and the onset of chronic kidney disease (CKD) [1].
Defining this tipping point has been a major challenge for a variety of technical reasons. But in a study just published in the journal Science Translational Medicine, researchers have discovered a molecular switch involved in controlling the transition from normal tissue repair to incomplete, or permanent, damage [2]. The NIH-supported researchers also suggest a possible drug candidate to control this switch and slow the progression of CKD.
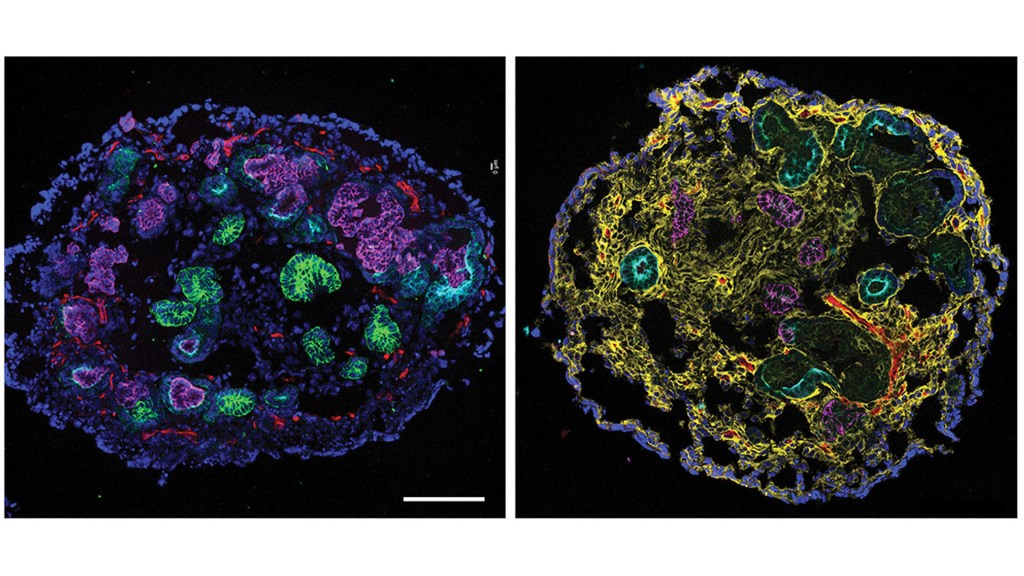
Also impressive is that the team broke through these longstanding technical problems without probing or testing a single person with CKD. They made their discovery using kidney organoids, or miniature human kidneys, that are grown in a lab dish and naturally model the repair process that takes place in our bodies.
The latest findings come from a team led by Ryuji Morizane, Massachusetts General Hospital and Harvard Medical School, Boston. The researchers recognized that earlier studies in animal models had identified processes involved in kidney injury and repair. But so far, there’s been limited success in translating those discoveries into clinical advances. That’s because many potential treatments that have appeared safe and effective in animal models have proven to be either damaging to the kidneys or ineffective when studied in humans.
To continue the search, the Morizane lab generated human kidney organoids from induced pluripotent stem cells (iPSCs) and other sources that include multiple essential renal tissue types. Using their tiny human kidneys, Morizane and colleagues, including first author Navin Gupta, sought the molecules responsible for the transition from complete to incomplete kidney repair.
The team repeatedly exposed kidney organoids to the cancer chemotherapy drug cisplatin, which can damage the kidneys as an unwanted side effect. Afterwards, examining single cells from the organoid, the researchers looked for underlying changes in gene activity associated with the transition from kidney repair to permanent kidney damage.
All told, their studies identified 159 genes in 29 different pathways that activate when kidneys fully repaired themselves. They found that many of those genes, including two called FANCD2 and RAD51, grew less active as kidney damage became irreversible. These genes encode proteins that are known to play a role in a process whereby cells repair broken strands of DNA.
Further study of stored biopsied kidney tissue from people with diabetic kidney disease, the most common cause of kidney failure, corroborated the organoid data tying a loss of FANCD2 activity to incomplete repair of kidney tissue. That’s encouraging because it suggests the new discoveries made in kidney organoids exposed to cisplatin may be relevant to people suffering from various forms of kidney injury.
One of the big advantages of organoid studies is the ability to rapidly screen for promising new drug candidates in the lab. And, indeed, the researchers found that a drug candidate called SCR7 helped to maintain FANCD2 and RAD51 activity in chemotherapy-injured organoids, preventing irreversible damage.
While much more study is needed, the findings suggest a potentially promising new way to prevent the kidneys from reaching their “tipping point” into permanent damage, CKD, and the risk for kidney failure. They also suggest that further studies in kidney organoids may lead to treatments targeting other kidney diseases.
These latest findings also highlight important progress in human tissue engineering, with implications for a wide range of conditions. In addition to making fundamental new biomedical discoveries as this new study has done, one of the great hopes of such efforts, including NIH’s National Center for Advancing Translational Sciences’ Tissue Chip for Drug Screening, is to improve predictions of whether new drug candidates will be safe or toxic in humans, speeding advances toward the most promising new therapies.
March happens to be National Kidney Month, and it’s especially important to raise awareness because 90 percent of people with CKD don’t even know they have it. So, if you or a loved one is at risk for CKD, be vigilant. Meanwhile, the work continues through studies like this one to find better leads to help control CKD.
References:
[1] Chronic kidney disease in the United States, 2021. Centers for Disease Control and Prevention.
[2] Modeling injury and repair in kidney organoids reveals that homologous recombination governs tubular intrinsic repair. Gupta N, Matsumoto T, Hiratsuka K, Garcia Saiz E, Galichon P, Miyoshi T, Susa K, Tatsumoto N, Yamashita M, Morizane R. Sci Transl Med. 2022 Mar 2;14(634):eabj4772
Links:
Chronic Kidney Disease (National Institute of Diabetes and Digestive and Kidney Diseases/NIH)
National Kidney Month 2022 (NIDDK)
Morizane Lab (Harvard Medical School, Boston, MA)
Tissue Chip for Screening (National Center for Advancing Translational Sciences/NIH)
NIH Support: National Institute of Diabetes and Digestive and Kidney Diseases; National Institute of Biomedical Imaging and Bioengineering; National Center for Advancing Translational Sciences
Partnership to Expand Effective Gene Therapies for Rare Diseases
Posted on by Dr. Francis Collins
Rare diseases aren’t so rare. Collectively, up to 30 million Americans, many of them children, are born with one of the approximately 7,000 known rare diseases. Most of these millions of people also share a common genetic feature: their diseases are caused by an alteration in a single gene.
Many of these alterations could theoretically be targeted with therapies designed to correct or replace the faulty gene. But there have been significant obstacles in realizing this dream. The science of gene therapy has been making real progress, but pursuing promising approaches all the way to clinical trials and gaining approval from the U.S. Food and Drug Administration (FDA) is still very difficult. Another challenge is economic: for the rarest of these conditions (which is most of them), the market is so small that most companies have no financial incentive to pursue them.
To overcome these obstacles and provide hope for those with rare diseases, we need a new way of doing things. One way to do things differently—and more efficiently—is the recently launched Bespoke Gene Therapy Consortium (BGTC). It is a bold partnership of NIH, the FDA, 10 pharmaceutical companies, several non-profit organizations, and the Foundation for the National Institutes of Health [1]. Its aim: optimize the gene therapy development process and help fill the significant unmet medical needs of people with rare diseases.
The BGTC, which is also part of NIH’s Accelerating Medicines Partnership® (AMP®), will enable the easier, faster, and cheaper pursuit of “bespoke” gene therapies, meaning made for a particular customer or user. The goal of the Consortium is to reduce the cost of gene therapy protocols and increase the likelihood of success, making it more attractive for companies to invest in rare diseases and bring treatments to patients who desperately need them.
Fortunately, there is already some precedent. The BGTC effort builds on a pilot project led by NIH’s National Center for Advancing Translational Sciences (NCATS) known as Platform Vector Gene Therapy (PaVe-GT). This pilot project has helped to develop adeno-associated viruses (AAVs), which are small benign viruses engineered in the lab to carry a therapeutic gene. They are commonly used in gene therapy-related clinical trials of rare diseases.
Since the launch of PaVe-GT two years ago, the project has helped to introduce greater efficiency to gene therapy trials for rare disease. It’s also offered a way to get around the standard one-disease-at-a-time approach to therapeutic development that has stymied progress in treating rare conditions.
The BGTC will now continue to advance in-depth understanding of basic AAV biology and develop better gene therapies for rare and also common diseases. The consortium aims to develop a standard set of analytic tests to improve the production and functional assessment of AAVs and therapeutic genes. Such tests will be broadly applicable and will bring the needed manufacturing efficiency required for developing gene therapies for very rare conditions.
The BGTC also will work toward bringing therapies sooner to individuals in need. To start, BGTC-funded research will support four to six clinical trials, each focused on a distinct rare disease. While the details haven’t yet been decided, these diseases are expected to be rare, single-gene diseases that lack gene therapies or commercial programs in development, despite having substantial groundwork in place to enable the rapid initiation of preclinical and clinical studies.
Through these trials, the BGTC will chart a path from studies in animal models of disease to human clinical trials that cuts years off the development process. This will include exploring methods to streamline regulatory requirements and processes for FDA approval of safe and effective gene therapies, including developing standardized approaches to preclinical testing.
This work promises to be a significant investment in helping people with rare diseases. The NIH and private partners will contribute approximately $76 million over five years to support BGTC-funded projects. This includes about $39.5 million from the participating NIH institutes and centers, pending availability of funds. The NCATS, which is NIH’s lead for BGTC, is expected to contribute approximately $8 million over five years.
Today, only two rare inherited conditions have FDA-approved gene therapies. The hope is this investment will raise that number and ultimately reduce the many significant challenges, including health care costs, faced by families that have a loved one with a rare disease. In fact, a recent study found that health care costs for people with a rare disease are three to five times greater than those for people without a rare disease [2]. These families need help, and BGTC offers an encouraging new way forward for them.
References:
[1] NIH, FDA and 15 private organizations join forces to increase effective gene therapies for rare diseases. NIH news release, October 27, 2021.
[2] The IDeaS initiative: pilot study to assess the impact of rare diseases on patients and healthcare systems. Tisdale, A., Cutillo, C.M., Nathan, R. et al. Orphanet J Rare Dis 16, 429 (2021).
Links:
FAQ About Rare Diseases (National Center for Advancing Translational Sciences/NIH)
Bespoke Gene Therapy Consortium (BGTC)
Platform Vector Gene Therapy (NCATS)
Accelerating Medicines Partnership® (AMP®) (NIH)
NIH Support: National Center for Advancing Translational Sciences; Eunice Kennedy Shriver National Institute of Child Health and Human Development; National Eye Institute; National Heart, Lung, and Blood Institute; National Human Genome Research Institute; National Institute of Arthritis and Musculoskeletal and Skin Diseases; National Institute of Dental and Craniofacial Research; National Institute of Mental Health; National Institute of Neurological Disorders and Stroke; National Institute on Deafness and Other Communication Disorders; and NIH’s BRAIN Initiative.
Decoding Heart-Brain Talk to Prevent Sudden Cardiac Deaths
Posted on by Dr. Francis Collins

As a cardiac electrophysiologist, Deeptankar DeMazumder has worked for years with people at risk for sudden cardiac arrest (SCA). Despite the latest medical advances, less than 10 percent of individuals stricken with an SCA will survive this highly dangerous condition in which irregular heart rhythms, or arrhythmias, cause the heart suddenly to stop beating.
In his role as a physician, DeMazumder keeps a tight focus on the electrical activity in their hearts, doing his best to prevent this potentially fatal event. In his other role, as a scientist at the University of Cincinnati College of Medicine, DeMazumder is also driven by a life-saving aspiration: finding ways to identify at-risk individuals with much greater accuracy than currently possible—and to develop better ways of protecting them from SCAs. He recently received a 2020 NIH Director’s New Innovator Award to pursue one of his promising ideas.
SCAs happen without warning and can cause death within minutes. Poor heart function and abnormal heart rhythms are important risk factors, but it’s not possible today to predict reliably who will have an SCA. However, doctors already routinely capture a wealth of information in electrical signals from the heart using electrocardiograms (ECGs). They also frequently use electroencephalograms (EEGs) to capture electrical activity in the brain.
DeMazumder’s innovative leap is to look at these heart and brain signals jointly, as well as in new ways, during sleep. According to the physician-scientist, sleep is a good time to search for SCA signatures in the electrical crosstalk between the heart and the brain because many other aspects of brain activity quiet down. He also thinks it’s important to pay special attention to what happens to the body’s electrical signals during sleep because most sudden cardiac deaths happen early in the waking hours, for reasons that aren’t well understood.
He has promising preliminary evidence from both animal models and humans suggesting that signatures within heart and brain signals are unique predictors of sudden death, even in people who appear healthy [1]. DeMazumder has already begun developing a set of artificial intelligence algorithms for jointly deciphering waveform signals from the heart, brain, and other body signals [2,3]. These new algorithms associate the waveform signals with a wealth of information available in electronic health records to improve upon the algorithm’s ability to predict catastrophic illness.
DeMazumder credits his curiosity about what he calls the “art and science of healing” to his early childhood experiences and his family’s dedication to community service in India. It taught him to appreciate the human condition, and he has integrated this life-long awareness into his Western medical training and his growing interest in computer science.
For centuries, humans have talked about how true flourishing needs both head and heart. In DeMazumder’s view, science is just beginning to understand the central role of heart-brain conversations in our health. As he continues to capture and interpret these conversations through his NIH-supported work, he hopes to find ways to identify individuals who don’t appear to have serious heart disease but may nevertheless be at high risk for SCA. In the meantime, he will continue to do all he can for the patients in his care.
References:
[1] Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Dey S, DeMazumder D, Sidor A, Foster DB, O’Rourke B. Circ Res. 2018;123(3):356-371.
[2] Entropy of cardiac repolarization predicts ventricular arrhythmias and mortality in patients receiving an implantable cardioverter-defibrillator for primary prevention of sudden death. DeMazumder D, Limpitikul WB, Dorante M, et al. Europace. 2016;18(12):1818-1828.
[3] Dynamic analysis of cardiac rhythms for discriminating atrial fibrillation from lethal ventricular arrhythmias. DeMazumder D, Lake DE, Cheng A, et al. Circ Arrhythm Electrophysiol. 2013;6(3):555-561.
Links:
Sudden Cardiac Arrest (National Heart, Lung, and Blood Institute/NIH)
Deeptankar DeMazumder (University of Cincinnati College of Medicine)
DeMazumder Project Information (NIH RePORTER)
NIH Director’s New Innovator Award (Common Fund)
NIH Support: National Heart, Lung, and Blood Institute; Common Fund
Tackling Cancer Metastasis with Engineered Blood Platelets
Posted on by Dr. Francis Collins

Credit: Dan Hixson/University of Utah College of Engineering, Salt Lake City
When cancer cells spread to new parts of the body in a process called metastasis, they often get there by traveling through the bloodstream. To avoid alerting the immune system and possibly triggering their demise, cancer cells coax circulating blood platelets to glom onto their surfaces and mask them from detection. This deceptive arrangement has raised a tantalizing possibility: What if blood platelets could be programmed to recognize and take out those metastasizing cancer cells?
Tara Deans, University of Utah, Salt Lake City, was recently awarded a 2019 NIH Director’s New Innovator Award to do exactly that. It’s an exciting opportunity for a researcher who stumbled onto this innovative strategy quite by accident.
Deans is a bioengineer and expert in designing synthetic gene circuits. These circuits consist of small collections of genetic “parts” that can be assembled and integrated to program cells to behave differently than their natural counterparts [1]. In her initial work, Deans got these specialized gene circuits to prompt blood-forming stem cells to mass-produce platelets in the lab.
But blood platelets are unusual cells. They’re packed with many proteins that help to repair small nicks in blood vessels and stop the bleeding when we’re injured. Blood platelets do so even though they lack a nucleus and DNA to encode and make any of the proteins. Their protein cargo is pre-packaged and comes strictly from the bone marrow cells, called megakaryocytes, that produce them.
Deans realized that engineering platelets might pose a rare opportunity. She could wire the needed circuitry into the blood-forming stem cells and engineer them to make any desired therapeutic proteins, which are then loaded into the blood platelets for their 8- to 10-day lifespan. She started out producing blood platelets that could safely carry functional replacement enzymes in people with certain rare metabolic disorders.
As this research progressed, Deans got some troubling personal news: A friend was diagnosed with a blood cancer. At the time, Deans didn’t know much about the diagnosis. But, in reading about her friend’s cancer, she learned how metastasizing tumor cells interact with platelets.
That’s when Deans had her “aha” moment: maybe the engineered platelets could also be put to work in preventing metastasizing tumor cells from spreading.
Now, with her New Innovator Award, Deans will pursue this novel approach by engineering platelets to carry potentially promising cancer-fighting proteins. In principle, they could be tailored to fight breast, lung, and various other cancer types. Ultimately, she hopes that platelets could be engineered to target and kill circulating cancer cells before they move into other tissues.
There’s plenty of research ahead to work out the details of targeting the circulating cancer cells and then testing them in animal models before this strategy could ever be attempted in people. But Deans is excited about the path forward, and thinks that platelets hold great promise to function as unique drug delivery devices. It has not escaped her notice that this approach could work not only for controlling the spread of cancer cells, but also in treating other medical conditions.
Reference:
[1] Genetic circuits to engineer tissues with alternative functions. Healy CP, Deans TL. J Biol Eng. 2019 May 3;13:39.
Links:
Metastatic Cancer (National Cancer Institute/NIH)
Deans Lab (University of Utah, Salt Lake City)
Deans Project Information (NIH RePORTER)
NIH Director’s New Innovator Award (Common Fund)
NIH Support: Common Fund; National Cancer Institute
Gene-Editing Advance Puts More Gene-Based Cures Within Reach
Posted on by Dr. Francis Collins
There’s been tremendous excitement recently about the potential of CRISPR and related gene-editing technologies for treating or even curing sickle cell disease (SCD), muscular dystrophy, HIV, and a wide range of other devastating conditions. Now comes word of another remarkable advance—called “prime editing”—that may bring us even closer to reaching that goal.
As groundbreaking as CRISPR/Cas9 has been for editing specific genes, the system has its limitations. The initial version is best suited for making a double-stranded break in DNA, followed by error-prone repair. The outcome is generally to knock out the target. That’s great if eliminating the target is the desired goal. But what if the goal is to fix a mutation by editing it back to the normal sequence?
The new prime editing system, which was described recently by NIH-funded researchers in the journal Nature, is revolutionary because it offers much greater control for making a wide range of precisely targeted edits to the DNA code, which consists of the four “letters” (actually chemical bases) A, C, G, and T [1].
Already, in tests involving human cells grown in the lab, the researchers have used prime editing to correct genetic mutations that cause two inherited diseases: SCD, a painful, life-threatening blood disorder, and Tay-Sachs disease, a fatal neurological disorder. What’s more, they say the versatility of their new gene-editing system means it can, in principle, correct about 89 percent of the more than 75,000 known genetic variants associated with human diseases.
In standard CRISPR, a scissor-like enzyme called Cas9 is used to cut all the way through both strands of the DNA molecule’s double helix. That usually results in the cell’s DNA repair apparatus inserting or deleting DNA letters at the site. As a result, CRISPR is extremely useful for disrupting genes and inserting or removing large DNA segments. However, it is difficult to use this system to make more subtle corrections to DNA, such as swapping a letter T for an A.
To expand the gene-editing toolbox, a research team led by David R. Liu, Broad Institute of MIT and Harvard, Cambridge, MA, previously developed a class of editing agents called base editors [2,3]. Instead of cutting DNA, base editors directly convert one DNA letter to another. However, base editing has limitations, too. It works well for correcting four of the most common single letter mutations in DNA. But at least so far, base editors haven’t been able to make eight other single letter changes, or fix extra or missing DNA letters.
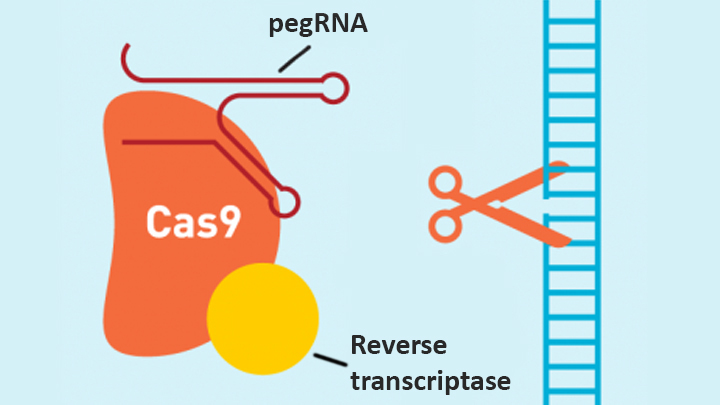
In contrast, the new prime editing system can precisely and efficiently swap any single letter of DNA for any other, and can make both deletions and insertions, at least up to a certain size. The system consists of a modified version of the Cas9 enzyme fused with another enzyme, called reverse transcriptase, and a specially engineered guide RNA, called pegRNA. The latter contains the desired gene edit and steers the needed editing apparatus to a specific site in a cell’s DNA.
Once at the site, the Cas9 nicks one strand of the double helix. Then, reverse transcriptase uses one DNA strand to “prime,” or initiate, the letter-by-letter transfer of new genetic information encoded in the pegRNA into the nicked spot, much like the search-and-replace function of word processing software. The process is then wrapped up when the prime editing system prompts the cell to remake the other DNA strand to match the new genetic information.
So far, in tests involving human cells grown in a lab dish, Liu and his colleagues have used prime editing to correct the most common mutation that causes SCD, converting a T to an A. They were also able to remove four DNA letters to correct the most common mutation underlying Tay-Sachs disease, a devastating condition that typically produces symptoms in children within the first year and leads to death by age four. The researchers also used their new system to insert new DNA segments up to 44 letters long and to remove segments at least 80 letters long.
Prime editing does have certain limitations. For example, 11 percent of known disease-causing variants result from changes in the number of gene copies, and it’s unclear if prime editing can insert or remove DNA that’s the size of full-length genes—which may contain up to 2.4 million letters.
It’s also worth noting that now-standard CRISPR editing and base editors have been tested far more thoroughly than prime editing in many different kinds of cells and animal models. These earlier editing technologies also may be more efficient for some purposes, so they will likely continue to play unique and useful roles in biomedicine.
As for prime editing, additional research is needed before we can consider launching human clinical trials. Among the areas that must be explored are this technology’s safety and efficacy in a wide range of cell types, and its potential for precisely and safely editing genes in targeted tissues within living animals and people.
Meanwhile, building on all these bold advances, efforts are already underway to accelerate the development of affordable, accessible gene-based cures for SCD and HIV on a global scale. Just last month, NIH and the Bill & Melinda Gates Foundation announced a collaboration that will invest at least $200 million over the next four years toward this goal. Last week, I had the chance to present this plan and discuss it with global health experts at the Grand Challenges meeting Addis Ababa, Ethiopia. The project is an unprecedented partnership designed to meet an unprecedented opportunity to address health conditions that once seemed out of reach but—as this new work helps to show—may now be within our grasp.
References:
[1] Search-and-replace genome editing without double-strand breaks or donor DNA. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, Liu DR. Nature. Online 2019 October 21. [Epub ahead of print]
[2] Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Nature. 2016 May 19;533(7603):420-424.
[3] Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR. Nature. 2017 Nov 23;551(7681):464-471.
Links:
Tay-Sachs Disease (Genetics Home Reference/National Library of Medicine/NIH)
Sickle Cell Disease (National Heart, Lung, and Blood Institute/NIH)
Cure Sickle Cell Initiative (NHLBI)
What are Genome Editing and CRISPR-Cas9? (National Library of Medicine/NIH)
Somatic Cell Genome Editing Program (Common Fund/NIH)
David R. Liu (Harvard, Cambridge, MA)
NIH Support: National Institute of Allergy and Infectious Diseases; National Human Genome Research Institute; National Institute for General Medical Sciences; National Institute of Biomedical Imaging and Bioengineering; National Center for Advancing Translational Sciences
Making Personalized Blood-Brain Barriers in a Dish
Posted on by Dr. Francis Collins

The blood-brain barrier, or BBB, is a dense sheet of cells that surrounds most of the brain’s blood vessels. The BBB’s tiny gaps let vital small molecules, such as oxygen and water, diffuse from the bloodstream into the brain while helping to keep out larger, impermeable foreign substances that don’t belong there.
But in people with certain neurological disorders—such as amyotrophic lateral sclerosis (ALS) and Huntington’s disease—abnormalities in this barrier may block the entry of biomolecules essential to healthy brain activity. The BBB also makes it difficult for needed therapies to reach their target in the brain.
To help look for solutions to these and other problems, researchers can now grow human blood-brain barriers on a chip like the one pictured above. The high-magnification image reveals some of the BBB’s cellular parts. There are endothelial-like cells (magenta), which are similar to those that line the small vessels surrounding the brain. In close association are supportive brain cells known as astrocytes (green), which help to regulate blood flow.
While similar organ chips have been created before, what sets apart this new BBB chip is its use of induced pluripotent stem cell (iPSC) technology combined with advanced chip engineering. The iPSCs, derived in this case from blood samples, make it possible to produce a living model of anyone’s unique BBB on demand.
The researchers, led by Clive Svendsen, Cedars-Sinai, Los Angeles, first use a biochemical recipe to coax a person’s white blood cells to become iPSCs. At this point, the iPSCs are capable of producing any other cell type. But the Svendsen team follows two different recipes to direct those iPSCs to differentiate into endothelial and neural cells needed to model the BBB.
Also making this BBB platform unique is its use of a sophisticated microfluidic chip, produced by Boston-based Emulate, Inc. The chip mimics conditions inside the human body, allowing the blood-brain barrier to function much as it would in a person.
The channels enable researchers to flow cerebral spinal fluid (CSF) through one side and blood through the other to create the fully functional model tissue. The BBB chips also show electrical resistance and permeability just as would be expected in a person. The model BBBs are even able to block the entry of certain drugs!
As described in Cell Stem Cell, the researchers have already created BBB chips using iPSCs from a person with Huntington’s disease and another from an individual with a rare congenital disorder called Allan-Herndon-Dudley syndrome, an inherited disorder of brain development.
In the near term, his team has plans to model ALS and Parkinson’s disease on the BBB chips. Because these chips hold the promise of modeling the human BBB more precisely than animal models, they may accelerate studies of potentially promising new drugs. Svendsen suggests that individuals with neurological conditions might one day have their own BBB chips made on demand to help in selecting the best-available therapeutic options for them. Now that’s a future we’d all like to see.
Reference:
[1] Human iPSC-Derived Blood-Brain Barrier Chips Enable Disease Modeling and Personalized Medicine Applications. Vatine GD, Barrile R, Workman MJ, Sances S, Barriga BK, Rahnama M, Barthakur S, Kasendra M, Lucchesi C, Kerns J, Wen N, Spivia WR, Chen Z, Van Eyk J, Svendsen CN. Cell Stem Cell. 2019 Jun 6;24(6):995-1005.e6.
Links:
Tissue Chip for Drug Screening (National Center for Advancing Translational Sciences/NIH)
Stem Cell Information (NIH)
Svendsen Lab (Cedars-Sinai, Los Angeles)
NIH Support: National Institute of Neurological Disorders and Stroke; National Center for Advancing Translational Sciences
Next Page