cancer
Microbe Normally Found in the Mouth May Drive Progression of Colorectal Cancer
Posted on by Dr. Monica M. Bertagnolli

Colorectal cancer is a leading cause of death from cancer in the United States. We know that risk of colorectal cancer goes up with age, certain coexisting health conditions, family history, smoking, alcohol use, and other factors. Researchers are also trying to learn more about what leads colorectal cancer to grow and spread. Now, findings from a new study supported in part by NIH add to evidence that colorectal tumor growth may be driven by a surprising bad actor: a microbe that’s normally found in the mouth.1
The findings, reported in Nature, suggest that a subtype of the bacterium Fusobacterium nucleatum has distinct genetic properties that may allow it to withstand acidic conditions in the stomach, infect colorectal tumors, and potentially drive their growth, which may lead to poorer patient outcomes. The discoveries suggest that the microbe could eventually be used as a target for detecting and treating colorectal cancer.
The study was conducted by a team led by Susan Bullman and Christopher D. Johnston at the Fred Hutchinson Cancer Center in Seattle. In 2022, the team published findings from a pair of studies implicating Fusobacterium nucleatum in the progression and spread of colorectal cancer.2,3 Their findings weren’t the first to suggest a link between the microbe and colorectal cancer. But their work offered important evidence that the microbe might alter colorectal tumors in ways that made them more likely to grow and spread. They also found that the microbe may affect the way colorectal cancer responds to or resists chemotherapy treatment.
Follow-up studies suggested there might be more to the story, pointing to the possibility that certain strains of the bacterium might differ from others in important ways. The findings suggested that there may be a more specific subtype, not yet defined, that was responsible for driving colorectal cancer growth.
To look deeper into this in the new study, Bullman and Johnston, with first author Martha Zepeda Rivera, analyzed a collection of 55 strains of the microbe taken from human colorectal cancer samples. They also compared these at the genetic level to another 80 strains of the microbe taken from the mouths of people who didn’t have cancer.
Their studies uncovered 483 genetic factors that turned up more often in Fusobacterium nucleatum from colorectal tumors. Those strains mainly belonged to a subspecies called Fusobacterium nucleatum animalis (Fna). More detailed study led to another surprise. The Fna included two genetically distinct groups or “clades” that had never been described, which the researchers called Fna C1 and C2. It turned out that only Fna C2 occurs at high levels in colorectal tumors.
The researchers found that this specific subtype within colorectal tumors carries 195 genetic factors that may allow it to grow more rapidly, withstand the acidic environment in the stomach, and take up residence in the gastrointestinal tract, where it can drive colorectal cancer growth. When the researchers infected a mouse model of colitis, a condition involving inflamed intestines that is a risk factor for colorectal cancer, they found that Fna C2 caused the development of more tumors compared to those infected with Fna C1.
Studies of tumors from 116 patients with colorectal cancer also showed more Fna C2. It was elevated in about 50% of cases. In fact, only this strain turned up more often in cancer compared to healthy tissue nearby. Stool samples of 627 people with colorectal cancer and 619 healthy people also showed more of this specific microbial strain in association with cancer.
This discovery is important because it suggests it’s only the Fna C2 subtype that’s associated with driving colorectal tumor growth, meaning it could help in the development of new methods for colorectal cancer screening and treatment. The researchers suggest it may one day even be possible to develop microbial-based therapies using modified versions of the bacterial strain to deliver treatments straight into tumors.
In addition, while the microbe is normally found in healthy mouths, it’s also enriched in periodontal (gum) disease, dental infections, and oral cancers.4 It will be interesting to learn more in future studies about the connections between various Fusobacterium nucleatum subtypes, oral health, and other health conditions throughout the body, including colorectal cancer.
References:
[1] Zepeda-Rivera M, et al. A distinct Fusobacterium nucleatum clade dominates the colorectal cancer niche. Nature. DOI: 10.1038/s41586-024-07182-w (2024).
[2] LaCourse KD, et al. The cancer chemotherapeutic 5-fluorouracil is a potent Fusobacterium nucleatum inhibitor and its activity is modified by intratumoral microbiota. Cell Rep. DOI: 10.1016/j.celrep.2022.111625 (2022).
[3] Galeano Niño JL, et al. Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature. DOI: 10.1038/s41586-022-05435-0 (2022).
[4] Chen Y, et al. More Than Just a Periodontal Pathogen –the Research Progress on Fusobacterium nucleatum. Front Cell Infect Microbiol. DOI: 10.3389/fcimb.2022.815318 (2022).
NIH Support: National Institute of Dental and Craniofacial Research, National Cancer Institute
Immune Checkpoint Discovery Has Implications for Treating Cancer and Autoimmune Diseases
Posted on by Dr. Monica M. Bertagnolli
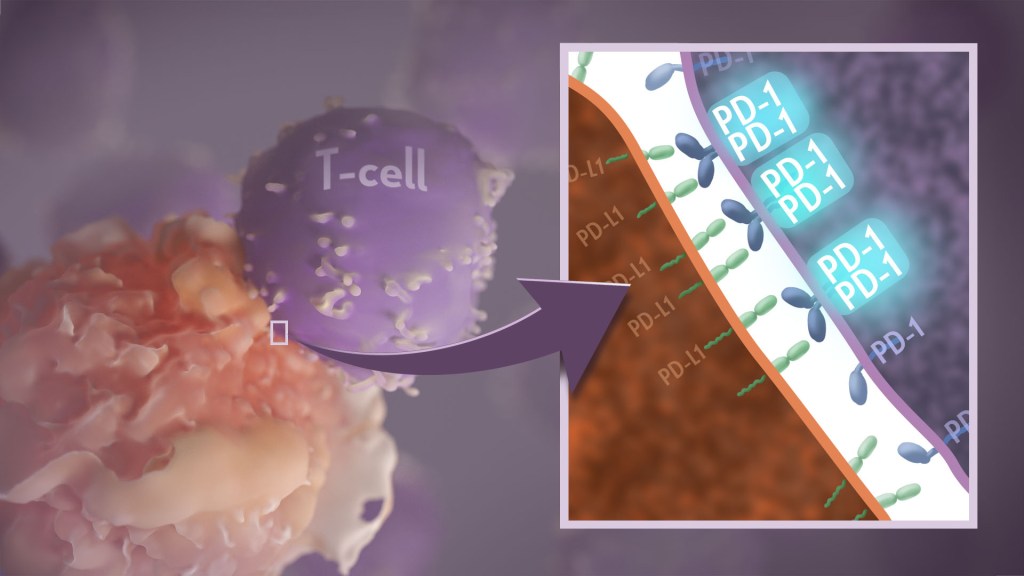
Your immune system should ideally recognize and attack infectious invaders and cancerous cells. But the system requires safety mechanisms, or brakes, to keep it from damaging healthy cells. To do this, T cells—the immune system’s most powerful attackers—rely on immune “checkpoints” to turn immune activation down when they receive the right signal. While these interactions have been well studied, a research team supported in part by NIH has made an unexpected discovery into how a key immune checkpoint works, with potentially important implications for therapies designed to boost or dampen immune activity to treat cancer and autoimmune diseases.1
The checkpoint in question is a protein called programmed cell death-1 (PD-1). Here’s how it works: PD-1 is a receptor on the surface of T cells, where it latches onto certain proteins, known as PD-L1 and PD-L2, on the surface of other cells in the body. When this interaction occurs, a signal is sent to the T cells that stops them from attacking these other cells.
Cancer cells often take advantage of this braking system, producing copious amounts of PD-L1 on their surface, allowing them to hide from T cells. An effective class of immunotherapy drugs used to treat many cancers works by blocking the interaction between PD-1 and PD-L1, to effectively release the brakes on the immune system to allow the T cells to unleash an assault on cancer cells. Researchers have also developed potential treatments for autoimmune diseases that take the opposite tact: stimulating PD-1 interaction to keep T cells inactive. These PD-1 “agonists” have shown promise in clinical trials as treatments for certain autoimmune diseases.
However, to fulfill the promise of these approaches for treating cancer and autoimmune diseases, a better understanding of precisely how PD-1 works to suppress T cell activity is still needed. The thinking has long been that individual PD-1 receptors act alone. But, as reported in Science Immunology, it turns out that this may not usually be the case. A team led by Jun Wang and Xiangpeng Kong of New York University Langone Health’s Perlmutter Cancer Center, with Elliot Philips of NYU and Michael Dustin of the University of Oxford, U.K., used sophisticated techniques to look for evidence of what happens when PD-1 proteins work together in pairs.
They found that PD-1’s tendency to link, or not link, with a second PD-1 protein to form what’s known as a “dimer” depends on interactions with portions of the protein that cross the immune cell membrane. They also found that, when PD-1 receptors pair up, they do a better job of squashing immune responses. The findings also showed that a single change in the amino acid structure in the portion of PD-1 that crosses the cell membrane can strengthen or weaken immune responses.
One reason why these fundamental discoveries are exciting is they suggest that interfering with PD-1’s ability to form dimers might make immunotherapy treatments for cancer more effective. In addition, treatments that strengthen interactions between paired PD-1 receptors might aid in the design of promising new drug classes that are intended to tamp down inflammation seen in people with some autoimmune diseases, including rheumatoid arthritis, lupus, and type 1 diabetes. The research team now plans to conduct further investigations of PD-1 blockers and agonists to explore whether these findings could eventually lead to more effective treatments for both cancer and autoimmune diseases.
Reference:
[1] Philips EA, et al. Transmembrane domain-driven PD-1 dimers mediate T cell inhibition. Science Immunology. DOI: 10.1126/sciimmunol.ade6256 (2024).
NIH Support: National Institute of Allergy and Infectious Diseases, National Cancer Institute, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institute of General Medical Sciences
A New Target to Improve the Health and Lives of Childhood Cancer Survivors: Diabetes Prevention
Posted on by Dr. Monica M. Bertagnolli
Before joining NIH, I conducted research on how inflammation drives colon cancer. I eventually led a trial to see if certain anti-inflammatory drugs could prevent the colon polyps that can can turn into cancer. The drugs worked; however, they also increased the risk of strokes and heart attacks, so they were not safe for people at high risk of cardiovascular disease.
The trial gave us valuable insight about the risks of these drugs and serves as an example of how clinicians and researchers must consider the needs of the whole patient rather than focusing on one organ system or disease. We have to recognize how certain interventions might improve one health issue but exacerbate another. This is especially important in adult survivors of childhood cancer. We know this population—about 500,000 people living in the U.S. according to 2020 estimates from the National Cancer Institute—faces an increased risk of developing chronic health conditions, including diabetes.
NIH supports numerous researchers working to understand better the health outcomes in childhood cancer survivors. One team at St. Jude Children’s Research Hospital in Memphis has been following more than 3,500 adults who had been diagnosed with childhood cancer. Known as the St. Jude LIFE cohort, the participants undergo regular health screenings and researchers use the information to determine the prevalence and predictors of health issues and to identify interventions to reduce risks.
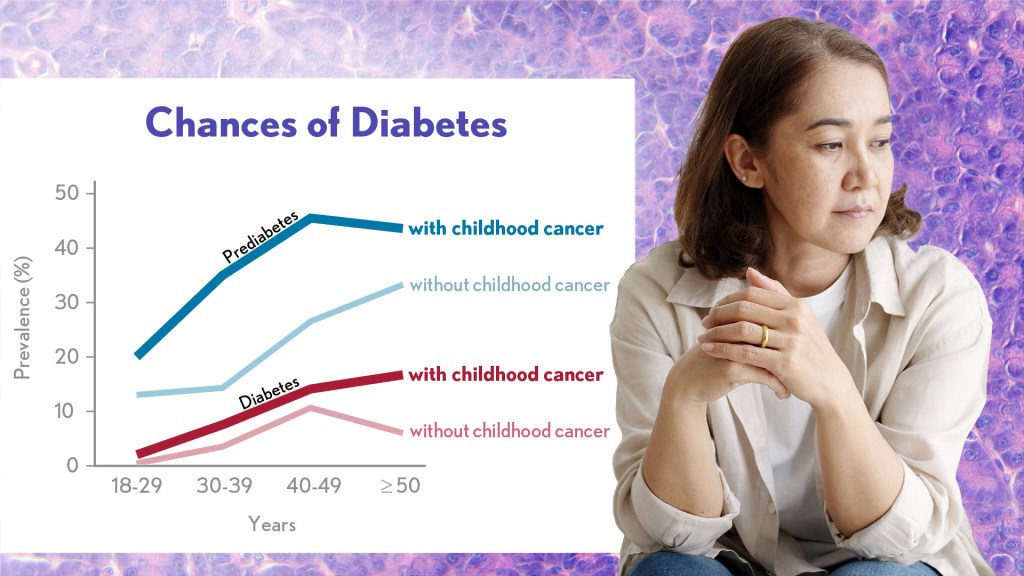
A new analysis published in the Journal of Clinical Oncology1 shows that prediabetes is highly prevalent in the St. Jude LIFE cohort: about one in every three survivors in the study had prediabetes by a median age of 30, compared to about one in five similarly aged adults without a cancer history. Prediabetes means that a person has higher than normal blood sugar levels but not high enough for a diagnosis of diabetes. Without intervention, many people with prediabetes will later develop diabetes.
By the time survivors in the study entered their 40s, more than half of them had prediabetes or diabetes, putting their future health at more risk compared to the general population. While these findings aren’t good news, they suggest that efforts to detect prediabetes and encourage lifestyle or treatment interventions before survivors go on to have more serious health complications can ensure that more people will live longer, healthier lives.
The research team, led by Stephanie Dixon, found that among 695 survivors with prediabetes who were followed over time, 10 percent progressed to diabetes within five years. The researchers also noted an association between radiation exposure to the pancreas and increased risk of prediabetes and diabetes. The pancreas produces insulin, and people are diagnosed with diabetes when the pancreas does not produce enough insulin to keep up with demand.
Their findings further suggest that, compared to survivors with normal blood sugar levels, those with prediabetes also had higher risk for future heart attack and chronic kidney disease. Those who progressed to diabetes also had more risk for developing stroke or cardiomyopathy (a condition where the heart pumps inefficiently and can lead to heart failure), in the future.
In the general population, prediabetes can be successfully managed through lifestyle changes, such as a healthy diet and exercise, as well as medication to prevent progression to diabetes and related health conditions. While this study is only a first step in identifying the consequences of prediabetes in survivors, it suggests that efforts to identify prediabetes and offer counseling on the importance of diabetes prevention may help more survivors of childhood cancers live long and healthy lives.
References:
[1] SB Dixon, et al. Prediabetes and Associated Risk of Cardiovascular Events and Chronic Kidney Disease Among Adult Survivors of Childhood Cancer in the St Jude Lifetime Cohort. Journal of Clinical Oncology DOI: 10.1200/JCO.23.01005 (2023).
NIH Support: National Cancer Institute
Turning Discoveries into Health for All
Posted on by Dr. Monica M. Bertagnolli

Greetings, blog readers! I’m Dr. Monica Bertagnolli, and I’m honored to be serving as the 17th Director of the National Institutes of Health. I’m excited to continue the NIH Director’s Blog to share with you the exciting discoveries and fascinating research conducted here at NIH and at the organizations we support in the U.S. and around the world. But before we start diving into the latest advances, I wanted to share a bit about myself and what I’m looking forward to as NIH Director.
I spent most of my career caring for people with cancer as a surgical oncologist and researcher before joining NIH last year as Director of the National Cancer Institute. While I miss the operating room (although I couldn’t stay away for long—more about that in a forthcoming blog!) and the opportunity to work with patients every day, I’m eager to serve the public in my new role as NIH Director.
When I was growing up, my family raised sheep and cattle on a ranch at the base of the Wind River Mountains in Wyoming. I know the health challenges that come with living in a rural area: Not everyone has access to an academic medical center and clinical studies, and managing the logistics of routine and preventive check-ups can be difficult. Unfortunately, many of our research advances are not reaching enough people in these areas.
As a cancer survivor, I am keenly aware that I’ve been fortunate to have access to excellent care, which has been directly informed by NIH-funded research over the past five decades. I know the transformative power of research to save lives, but from my experience as a clinician, I know that it is not always possible for people to receive the care that they need due to financial, geographic, or cultural barriers. It is unacceptable for the benefits of NIH-funded biomedical research to be available to some but not all.
That’s why one of my goals as NIH Director is to ensure the biomedical research enterprise and its discoveries—from basic to clinical research—are more inclusive and accessible to people from all walks of life, including rural areas. Income, age, race, ethnicity, geographic location, and disability status should not be barriers to participating in research or to benefitting from research advances. By meeting people where they are and engaging more communities as our research partners, I believe we can also make significant progress in rebuilding trust in science across the country.
Right now, we have an unprecedented opportunity to embrace and increase access to innovation: Our knowledge and technology have developed to the point that we should be able to deliver evidence-based, data-driven health care to everyone. This is an exciting time for science, and I can’t wait to share more with you in the weeks and months to come.
Persistence Pays Off: Recognizing Katalin Karikó and Drew Weissman, the 2023 Nobel Prize Winners in Physiology or Medicine
Posted on by Lawrence Tabak, D.D.S., Ph.D.
Last week, biochemist Katalin Karikó and immunologist Drew Weissman earned the Nobel Prize in Physiology or Medicine for their discoveries that enabled the development of effective messenger RNA (mRNA) vaccines against COVID-19. On behalf of the NIH community, I’d like to congratulate Karikó and Weissman and thank them for their persistence in pursuing their investigations. NIH is proud to have supported their seminal research, cited by the Nobel Assembly as key publications.1,2,3
While the lifesaving benefits of mRNA vaccines are now clearly realized, Karikó and Weissman’s breakthrough finding in 2005 was not fully appreciated at the time as to why it would be significant. However, their dogged dedication to gaining a better understanding of how RNA interacts with the immune system underscores the often-underappreciated importance of incremental research. Following where the science leads through step-by-step investigations often doesn’t appear to be flashy, but it can end up leading to major advances.
To best describe Karikó and Weissman’s discovery, I’ll first do a quick review of vaccine history. As many of you know, vaccines stimulate our immune systems to protect us from getting infected or from getting very sick from a specific pathogen. Since the late 1700s, scientists have used various approaches to design effective vaccines. Some vaccines introduce a weakened or noninfectious version of a virus to the body, while others present only a small part of the virus, like a protein. The immune system detects the weak or partial virus and develops specialized defenses against it. These defenses work to protect us if we are ever exposed to the real virus.
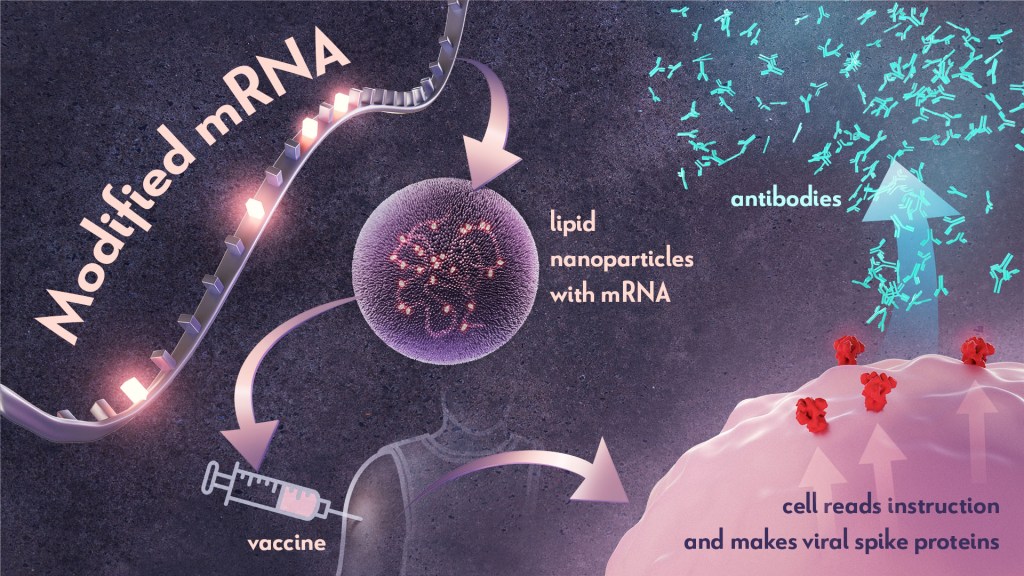
In the early 1990s, scientists began exploring a different approach to vaccines that involved delivering genetic material, or instructions, so the body’s own cells could make the virus proteins that stimulate an immune response.4,5 Because this approach eliminates the step of growing virus or virus protein in the laboratory—which can be difficult to do in very large quantities and can require a lot of time and money—it had potential, in theory, to be a faster and cheaper way to manufacture vaccines.
Scientists were exploring two types of vaccines as part of this new approach: DNA vaccines and messenger RNA (mRNA) vaccines. DNA vaccines deliver an encoded protein recipe that the cell first copies or transcribes before it starts making protein. For mRNA vaccines, the transcription process is done in the laboratory, and the vaccine delivers the “readable” instructions to the cell for making protein. However, mRNA was not immediately a practical vaccine approach due to several scientific hurdles, including that it caused inflammatory reactions that could be unhealthy for people.
Unfazed by the challenges, Karikó and Weissman spent years pursuing research on RNA and the immune system. They had a brilliant idea that they turned into a significant discovery in 2005 when they proved that inserting subtle chemical modifications to lab-transcribed mRNA eliminated the unwanted inflammatory response.1 In later studies, the pair showed that these chemical modifications also increased protein production.2,3 Both discoveries would be critical to advancing the use of mRNA-based vaccines and therapies.
Earlier theories that mRNA could enable rapid vaccine development turned out to be true. By March 2020, the first clinical trial of an mRNA vaccine for COVID-19 had begun enrolling volunteers, and by December 2020, health care workers were receiving their first shots. This unprecedented timeline was only possible because of Karikó and Weissman’s decades of work, combined with the tireless efforts of many academic, industry and government scientists, including several from the NIH intramural program. Now, researchers are exploring how mRNA could be used in vaccines for other infectious diseases and in cancer vaccines.
As an investigator myself, I’m fascinated by how science continues to build on itself—a process that is done out of the public eye. Luckily every year, the Nobel Prize briefly illuminates for the larger public this long arc of scientific discovery. The Nobel Assembly’s recognition of Karikó and Weissman is a tribute to all scientists who do the painstaking work of trying to understand how things work. Many of the tools we have today to better prevent and treat diseases would not have been possible without the brilliance, tenacity and grit of researchers like Karikó and Weissman.
References:
- K Karikó, et al. Suppression of RNA Recognition by Toll-like Receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity DOI: 10.1016/j.immuni.2005.06.008 (2005).
- K Karikó, et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Molecular Therapy DOI: 10.1038/mt.2008.200 (2008).
- BR Anderson, et al. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Research DOI: 10.1093/nar/gkq347 (2010).
- DC Tang, et al. Genetic immunization is a simple method for eliciting an immune response. Nature DOI: 10.1038/356152a0 (1992).
- F Martinon, et al. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. European Journal of Immunology DOI: 10.1002/eji.1830230749 (1993).
NIH Support:
Katalin Karikó: National Heart, Lung, and Blood Institute; National Institute of Neurological Disorders and Stroke
Drew Weissman: National Institute of Allergy and Infectious Diseases; National Institute of Dental and Craniofacial Research; National Heart, Lung, and Blood Institute
Rice-Sized Device Tests Brain Tumor’s Drug Responses During Surgery
Posted on by Lawrence Tabak, D.D.S., Ph.D.

Scientists have made remarkable progress in understanding the underlying changes that make cancer grow and have applied this knowledge to develop and guide targeted treatment approaches to vastly improve outcomes for people with many cancer types. And yet treatment progress for people with brain tumors known as gliomas—including the most aggressive glioblastomas—has remained slow. One reason is that doctors lack tests that reliably predict which among many therapeutic options will work best for a given tumor.
Now an NIH-funded team has developed a miniature device with the potential to change this for the approximately 25,000 people diagnosed with brain cancers in the U.S. each year [1]. When implanted into cancerous brain tissue during surgery, the rice-sized drug-releasing device can simultaneously conduct experiments to measure a tumor’s response to more than a dozen drugs or drug combinations. What’s more, a small clinical trial reported in Science Translational Medicine offers the first evidence in people with gliomas that these devices can safely offer unprecedented insight into tumor-specific drug responses [2].
These latest findings come from a Brigham and Women’s Hospital, Boston, team led by Pierpaolo Peruzzi and Oliver Jonas. They recognized that drug-screening studies conducted in cells or tissue samples in the lab too often failed to match what happens in people with gliomas undergoing cancer treatment. Wide variation within individual brain tumors also makes it hard to predict a tumor’s likely response to various treatment options.
It led them to an intriguing idea: Why not test various therapeutic options in each patient’s tumor? To do it, they developed a device, about six millimeters long, that can be inserted into a brain tumor during surgery to deliver tiny doses of up to 20 drugs. Doctors can then remove and examine the drug-exposed cancerous tissue in the laboratory to determine each treatment’s effects. The data can then be used to guide subsequent treatment decisions, according to the researchers.
In the current study, the researchers tested their device on six study volunteers undergoing brain surgery to remove a glioma tumor. For each volunteer, the device was implanted into the tumor and remained in place for about two to three hours while surgeons worked to remove most of the tumor. Next, the device was taken out along with the last piece of a tumor at the end of the surgery for further study of drug responses.
Importantly, none of the study participants experienced any adverse effects from the device. Using the devices, the researchers collected valuable data, including how a tumor’s response changed with varying drug concentrations or how each treatment led to molecular changes in the cancerous cells.
More research is needed to better understand how use of such a device might change treatment and patient outcomes in the longer term. The researchers note that it would take more than a couple of hours to determine how treatments produce less immediate changes, such as immune responses. As such, they’re now conducting a follow-up trial to test a possible two-stage procedure, in which their device is inserted first using minimally invasive surgery 72 hours prior to a planned surgery, allowing longer exposure of tumor tissue to drugs prior to a tumor’s surgical removal.
Many questions remain as they continue to optimize this approach. However, it’s clear that such a device gives new meaning to personalized cancer treatment, with great potential to improve outcomes for people living with hard-to-treat gliomas.
References:
[1] National Cancer Institute Surveillance, Epidemiology, and End Results Program. Cancer Stat Facts: Brain and Other Nervous System Cancer.
[2] Peruzzi P et al. Intratumoral drug-releasing microdevices allow in situ high-throughput pharmaco phenotyping in patients with gliomas. Science Translational Medicine DOI: 10.1126/scitranslmed.adi0069 (2023).
Links:
Brain Tumors – Patient Version (National Cancer Institute/NIH)
Pierpaolo Peruzzi (Brigham and Women’s Hospital, Boston, MA)
Jonas Lab (Brigham and Women’s Hospital, Boston, MA)
NIH Support: National Cancer Institute, National Institute of Biomedical Imaging and Bioengineering, National Institute of Neurological Disorders and Stroke
New Approach to ‘Liquid Biopsy’ Relies on Repetitive RNA in the Bloodstream
Posted on by Lawrence Tabak, D.D.S., Ph.D.

It’s always best to diagnose cancer at an early stage when treatment is most likely to succeed. Unfortunately, far too many cancers are still detected only after cancer cells have escaped from a primary tumor and spread to distant parts of the body. This explains why there’s been so much effort in recent years to develop liquid biopsies, which are tests that can pick up on circulating cancer cells or molecular signs of cancer in blood or other bodily fluids and reliably trace them back to the organ in which a potentially life-threatening tumor is growing.
Earlier methods to develop liquid biopsies for detecting cancers often have relied on the presence of cancer-related proteins and/or DNA in the bloodstream. Now, an NIH-supported research team has encouraging evidence to suggest that this general approach to detecting cancers—including aggressive pancreatic cancers—may work even better by taking advantage of signals from a lesser-known form of genetic material called noncoding RNA.
The findings reported in Nature Biomedical Engineering suggest that the new liquid biopsy approach may aid in the diagnosis of many forms of cancer [1]. The studies show that the sensitivity of the tests varies—a highly sensitive test is one that rarely misses cases of disease. However, they already have evidence that millions of circulating RNA molecules may hold promise for detecting cancers of the liver, esophagus, colon, stomach, and lung.
How does it work? The human genome contains about 3 billion paired DNA letters. Most of those letters are transcribed, or copied, into single-stranded RNA molecules. While RNA is best known for encoding proteins that do the work of the cell, most RNA never gets translated into proteins at all. This noncoding RNA includes repetitive RNA that can be transcribed from millions of repeat elements—patterns of the same few DNA letters occurring multiple times in the genome.
Common approaches to studying RNA don’t analyze repetitive RNA, so its usefulness as a diagnostic tool has been unclear—until recently. Last year, the lab of Daniel Kim at the University of California, Santa Cruz reported [2] that a key genetic mutation that occurs early on in some cancers causes repetitive RNA molecules to be secreted in large quantities from cancer cells, even at the earliest stages of cancer. Non-cancerous cells, by comparison, release much less repetitive RNA.
The findings suggested that liquid biopsy tests that look for this repetitive, noncoding RNA might offer a powerful new way to detect cancers sooner, according to the authors. But first they needed a method capable of measuring it. Due to its oftentimes uncertain functions, the researchers have referred to repetitive, noncoding RNA as “dark matter.”
Using a liquid biopsy platform they developed called COMPLETE-seq, Kim’s team trained computers to detect cancers by looking for patterns in RNA data. The platform enables sequencing and analysis of all protein coding and noncoding RNAs—including any RNA from more than 5 million repeat elements—present in a blood sample. They found that their classifiers worked better when repetitive RNAs were included. The findings lend support to the idea that repetitive, noncoding RNA in the bloodstream is a rich source of information for detecting cancers, which has previously been overlooked.
In a study comparing blood samples from healthy people to those with pancreatic cancer, the COMPLETE-seq technology showed that nearly all people in the study with pancreatic cancer had more repetitive, noncoding RNA in their blood samples compared to healthy people, according to the researchers. They used the COMPLETE-seq test on blood samples from people with other types of cancer as well. For example, their test accurately detected 91% of colorectal cancer samples and 93% of lung cancer samples.
They now plan to look at many more cancer types with samples from additional patients representing a broad range of cancer stages. The goal is to develop a single RNA liquid biopsy test that could detect multiple forms of cancer with a high degree of accuracy and specificity. They note that such a test might also be used to guide treatment decisions and more readily detect a cancer’s recurrence. The hope is that one day a comprehensive liquid biopsy test including coding and noncoding RNA will catch many more cancers sooner, when treatment can be most successful.
References:
[1] RE Reggiardo et al. Profiling of repetitive RNA sequences in the blood plasma of patients with cancer. Nature Biomedical Engineering DOI: 10.1038/s41551-023-01081-7 (2023).
[2] RE Reggiardo et al. Mutant KRAS regulates transposable element RNA and innate immunity via KRAB zinc-finger genes. Cell Reports DOI: 10.1016/j.celrep.2022.111104 (2022).
Links:
Daniel Kim Lab (UC Santa Cruz)
Cancer Screening Overview (National Cancer Institute/NIH)
Early Detection (National Cancer Institute/NIH)
NIH Support: National Cancer Institute, National Heart, Lung, and Blood Institute, National Institute of Diabetes and Digestive and Kidney Diseases
Plans for New Cancer Center in Kansas
Posted on by Lawrence Tabak, D.D.S., Ph.D.

That afternoon, I gathered with some of the event participants for this photo. Starting in the back row (l-r), you see Senator Moran; Rachel Pepper, chief nursing officer, Kansas City Division, The University of Kansas Health System; Roy Jensen, director, The University of Kansas Cancer Center, Kansas City; Bob Page, president and CEO, The University of Kansas Health System, Kansas City. In the front row (l-r) are Joseph McGuirk, Division of Hematologic Malignancies and Cellular Therapeutics, University of Kansas Cancer Center, Kansas City; Britany Leiker, nurse manager, The University of Kansas Medical Center, Kansas City; Tammy Peterman, president, Kansas City Division, The University of Kansas Health System; Doug Girod, chancellor, University of Kansas, Lawrence; and I’m next standing at the end of the row.
The event was held on June 27 at The University of Kansas Health Education Building, Kansas City. Credit: Elissa Monroe, The University of Kansas Medical Center, Kansas City.
Basic Researchers Discover Possible Target for Treating Brain Cancer
Posted on by Lawrence Tabak, D.D.S., Ph.D.
Over the years, cancer researchers have uncovered many of the tricks that tumors use to fuel their growth and evade detection by the body’s immune system. More tricks await discovery, and finding them will be key in learning to target the right treatments to the right cancers.
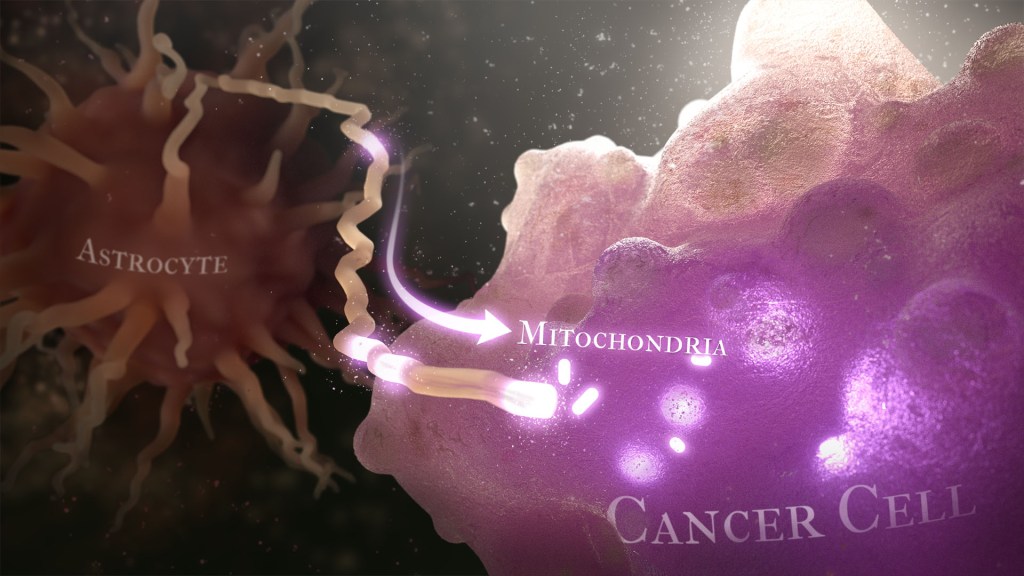
Recently, a team of researchers demonstrated in lab studies a surprising trick pulled off by cells from a common form of brain cancer called glioblastoma. The researchers found that glioblastoma cells steal mitochondria, the power plants of our cells, from other cells in the central nervous system [1].
Why would cancer cells do this? How do they pull it off? The researchers don’t have all the answers yet. But glioblastoma arises from abnormal astrocytes, a particular type of the glial cell, a common cell in the brain and spinal cord. It seems from their initial work that stealing mitochondria from neighboring normal cells help these transformed glioblastoma cells to ramp up their growth. This trick might also help to explain why glioblastoma is one of the most aggressive forms of primary brain cancer, with limited treatment options.
In the new study, published in the journal Nature Cancer, a team co-led by Justin Lathia, Lerner Research Institute, Cleveland Clinic, OH, and Hrvoje Miletic, University of Bergen, Norway, had noticed some earlier studies suggesting that glioblastoma cells might steal mitochondria. They wanted to take a closer look.
This very notion highlights an emerging and much more dynamic view of mitochondria. Scientists used to think that mitochondria—which can number in the thousands within a single cell—generally just stayed put. But recent research has established that mitochondria can move around within a cell. They sometimes also get passed from one cell to another.
It also turns out that the intercellular movement of mitochondria has many implications for health. For instance, the transfer of mitochondria helps to rescue damaged tissues in the central nervous system, heart, and respiratory system. But, in other circumstances, this process may possibly come to the rescue of cancer cells.
While Lathia, Miletic, and team knew that mitochondrial transfer was possible, they didn’t know how relevant or dangerous it might be in brain cancers. To find out, they studied mice implanted with glioblastoma tumors from other mice or people with glioblastoma. This mouse model also had been modified to allow the researchers to trace the movement of mitochondria.
Their studies show that healthy cells often transfer some of their mitochondria to glioblastoma cells. They also determined that those mitochondria often came from healthy astrocytes, a process that had been seen before in the recovery from a stroke.
But the transfer process isn’t easy. It requires that a cell expend a lot of energy to form actin filaments that contract to pull the mitochondria along. They also found that the process depends on growth-associated protein 43 (GAP43), suggesting that future treatments aimed at this protein might help to thwart the process.
Their studies also show that, after acquiring extra mitochondria, glioblastoma cells shift into higher gear. The cancerous cells begin burning more energy as their metabolic pathways show increased activity. These changes allow for more rapid and aggressive growth. Overall, the findings show that this interaction between healthy and cancerous cells may partly explain why glioblastomas are so often hard to beat.
While more study is needed to confirm the role of this process in people with glioblastoma, the findings are an important reminder that treatment advances in oncology may come not only from study of the cancer itself but also by carefully considering the larger context and environments in which tumors grow. The hope is that these intriguing new findings will one day lead to new treatment options for the approximately 13,000 people in the U.S. alone who are diagnosed with glioblastoma each year [2].
References:
[1] GAP43-dependent mitochondria transfer from astrocytes enhances glioblastoma tumorigenicity. Watson DC, Bayik D, Storevik S, Moreino SS, Hjelmeland AB, Hossain JA, Miletic H, Lathia JD et al. Nat Cancer. 2023 May 11. [Published online ahead of print.]
[2] CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011-2015. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. 2018 Oct 1, Neuro Oncol., p. 20(suppl_4):iv1-iv86.
Links:
Glioblastoma (National Center for Advancing Translational Sciences/NIH)
Brain Tumors (National Cancer Institute/NIH)
Justin Lathia Lab (Cleveland Clinic, OH)
Hrvoje Miletic (University of Bergen, Norway)
NIH Support: National Institute of Neurological Disorders and Stroke; National Center for Advancing Translational Sciences; National Cancer Institute; National Institute of Allergy and Infectious Diseases
Study Reveals How Epstein-Barr Virus May Lead to Cancer
Posted on by Lawrence Tabak, D.D.S., Ph.D.
Chances are good that you’ve had an Epstein-Barr virus (EBV) infection, usually during childhood. More than 90 percent of us have, though we often don’t know it. That’s because most EBV infections are mild or produce no symptoms at all.
But in some people, EBV can lead to other health problems. The virus can cause infectious mononucleosis (“mono”), type 1 diabetes, and other ailments. It also can persist in our bodies for years and cause increased risk later in life for certain cancers, such as lymphoma, leukemia, and head and neck cancer. Now, an NIH-funded team has some of the best evidence yet to explain how this EBV that hangs around may lead to cancer [1].
The paper, published recently in the journal Nature, shows that a key viral protein readily binds to a particular spot on a particular human chromosome. Where the protein accumulates, the chromosome becomes more prone to breaking for reasons that aren’t yet fully known. What the study makes clearer is that the breakage produces latently infected cells that are more likely over time to become cancerous.
This discovery paves the way potentially for ways to screen for and identify those at particular risk for developing EBV-associated cancers. It may also fuel the development of promising new ways to prevent these cancers from arising in the first place.
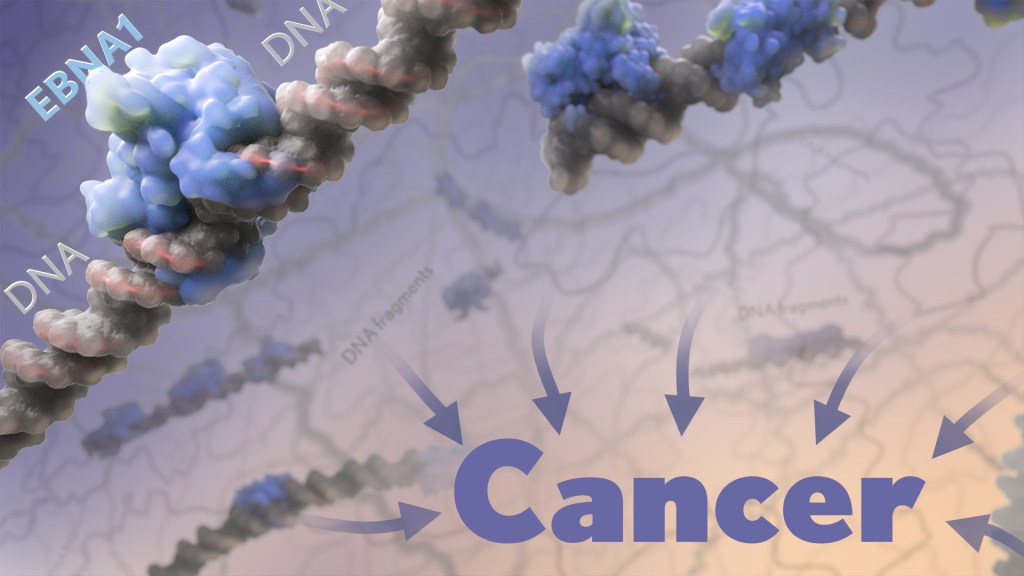
The work comes from a team led by Don Cleveland and Julia Su Zhou Li, University of California San Diego’s Ludwig Cancer Research, La Jolla, CA. Over the years, it’s been established that EBV, a type of herpes virus, often is detected in certain cancers, particularly in people with a long-term latent infection. What interested the team is a viral protein, called EBNA1, which routinely turns up in those same EBV-related cancers.
The EBNA1 protein is especially interesting because it binds viral DNA in particular spots, which allows the virus to persist and make more copies of itself. This discovery raised the intriguing possibility that the protein may also bind similar sequences in human DNA. While it had been suggested previously that this interaction might play a role in EBV-associated cancers, the details had remained murky—until now.
In the new study, the researchers first made uninfected human cells produce the viral EBNA1 protein. They then peered inside them with a microscope to see where those proteins went. In both healthy and cancerous human cells, they watched as EBNA1 proteins built up at two distinct spots and confirmed that this accumulation was dependent on the protein’s ability to bind DNA.
Next, they mapped where exactly EBNA1 binds to human DNA. Interestingly, it was along a repetitive non-protein-coding stretch of DNA on human chromosome 11. This region includes more than 300 copies of an 18-letter sequence that looks quite similar to the EBNA1-binding sites in its own viral genome.
What’s more, the researchers noticed that the repetitive DNA there takes on a structure that’s known for being unstable. And these so-called fragile sites are inherently prone to breaking.
The team went on to uncover evidence that the buildup of EBNA1 at this already fragile site only makes matters worse. In EBV-infected cells, increasing the amount of EBNA1 protein led to more chromosome 11 breaks. Those breaks showed up within a single day in about 40 percent of cells.
For these cells, those breaks also may be a double whammy. That’s because the breaks are located next to neighboring genes with long recognized roles in regulating cell growth. When altered, these genes can contribute to turning a cell cancerous.
To further nail down the link to cancer, the researchers looked to whole-genome sequencing data for more than 2,400 cancers including 38 tumor types from the international Pan-Cancer Analysis of Whole Genomes consortium [2]. They found that tumors with detectable EBV also had an unusually high number of chromosome 11 abnormalities. In fact, that was true in every single case of head and neck cancer.
The findings suggest that people will vary in their susceptibility to EBNA1-induced DNA breaks along chromosome 11 based on the amount of EBNA1 protein in their latently infected cells. It also will depend on the number of EBV-like DNA repeats present in their DNA.
Given these new findings, it’s worth noting that the presence of EBV and the very same viral protein has been implicated also in the link between EBV and multiple sclerosis (MS) [3]. Together, these recent findings are a reminder of the value in pursuing an EBV vaccine that might thwart this infection and its associated conditions, including certain cancers and MS. And, we’re getting there. In fact, an early-stage clinical trial for an experimental EBV vaccine is now ongoing here at the NIH Clinical Center.
References:
[1] Chromosomal fragile site breakage by EBV-encoded EBNA1 at clustered repeats. Li JSZ, Abbasi A, Kim DH, Lippman SM, Alexandrov LB, Cleveland DW. Nature. 2023 Apr 12.
[2] Pan-cancer analysis of whole genomes. ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Nature.2020 Feb;578(7793):82-93.
[3] Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Lanz TV, Brewer RC, Steinman L, Robinson WH, et al. Nature. 2022 Mar;603(7900):321-327.
Links:
About Epstein-Barr Virus (Centers for Disease Control and Prevention, Atlanta)
Head and Neck Cancer (National Cancer Institute,/NIH)
Multiple Sclerosis (National Institute of Neurological Disorders and Stroke/NIH)
Don W. Cleveland Lab (University of California San Diego, La Jolla, CA)
NIH Support: National Institute of General Medical Sciences; National Institute of Environmental Health Sciences; National Cancer Institute
Next Page