rare disease
Unraveling the Role of the Skin Microbiome in Health and Disease
Posted on by Lindsey A. Criswell, M.D., M.P.H., D.Sc., National Institute of Arthritis and Musculoskeletal and Skin Diseases

Human skin is home to diverse ecosystems including bacteria, viruses, and fungi. These microbial communities comprise hundreds of species and are collectively known as the skin microbiome. The skin microbiome is thought to play a vital role in fending off disease-causing microorganisms (pathogens), boosting barrier protection, and aiding immune defenses.
Maintaining a balanced skin microbiome involves a complex and dynamic interplay among microorganisms, immune cells, skin cells, and other factors. In general, bacteria far outnumber viral, fungal, or other microbial species on the skin. Bacterial communities, which are strongly influenced by conditions such as skin moisture, temperature, and pH, vary widely across the body. For example, facial cheek skin hosts mostly Cutibacterium along with a bit of the skin fungus Malassezia. The heel is colonized by different types of bacteria including Staphylococcus and Corynebacteria.
In some diseases, such as acne and eczema, the skin microbiome is altered. Typically, this means an increase in pathogenic microorganisms and a decrease in beneficial ones. An altered skin microbiome can also be associated with inflammation, severe disease symptoms, and changes in the human immune system.
Heidi H. Kong is working to understand the role of the skin microbiome in health and disease. She is a senior investigator in the Intramural Research Program at NIH’s National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) and an adjunct investigator at NIH’s National Cancer Institute (NCI).
More than a decade ago, Kong and Julie A. Segre, an intramural researcher at NIH’s National Human Genome Research Institute, analyzed the microbial makeup of healthy individuals. Kong swabbed the skin of these healthy volunteers in 20 different sites, from the forehead to the toenail. The study revealed that the surface of the human body provides various environmental niches, depending on whether the skin is moist, dry, or sebaceous (oily). Different bacterial species predominate in each niche. Kong and Segre were particularly interested in body areas that have predilections for disease. For example, psoriasis is often found on the outside of elbows and knees, and the back of the scalp.
Earlier this year, Kong and Segre published another broad analysis of the human skin microbiome [1] in collaboration with scientists at the European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL-EBI), United Kingdom. This new catalog, called the Skin Microbial Genome Collection, is thought to identify about 85 percent of the microorganisms present on healthy skin from 19 body sites. It documents more than 600 bacterial species—including 174 that were discovered during the study—as well as more than 6,900 viruses and some fungi, including three newly discovered species.
Kong’s work has provided compelling evidence that the human immune system plays a role in shaping the skin microbiome. In 2018, she, Segre, and colleagues from the intramural programs of NCI and NIH’s National Institute of Allergy and Infectious Diseases analyzed skin from eight different sites on 27 people with a rare primary immunodeficiency disease known as DOCK8 deficiency [2].
People with the condition have recurrent infections in the skin, sinuses, and airways, and are susceptible to different cancers. Kong and colleagues found that the skin of people with DOCK8 deficiency contains significantly more DNA viruses (90 percent of the skin microbiome on average) than people without the condition (6 or 7 percent of the skin microbiome).
Other researchers are hoping to leverage features of the microbiome to develop targeted therapies for skin diseases. Richard L. Gallo, a NIAMS grantee at the University of California, San Diego, is currently focused on acne and eczema (also called atopic dermatitis). Acne is associated with certain strains of Cutibacterium acnes (C. acnes, formerly called Propionibacterium acnes or P. acnes). Eczema is often associated with Staphylococcus aureus (S. aureus).
Severe cases of acne and eczema are commonly treated with broad-spectrum antibiotics, which wipe out most of the bacteria, including beneficial species. The goal of microbiome-targeted therapy is to kill only the disease-associated bacteria and avoid increasing the risk that some strains will develop antibiotic resistance.
In 2020, Gallo and colleagues identified a strain of Staphylococcus capitis from healthy human skin (S. capitis E12) that selectively inhibits the growth of C. acnes without negatively impacting other bacteria or human skin cells [3]. S. capitis E12 produces four different toxins that act together to target C. acnes. The research team created an extract of the four toxins and tested it using animal models. In most cases, the extract was more potent at killing C. acnes—including acne-associated strains—than several commonly prescribed antibiotics (erythromycin, tetracycline, and clindamycin). And, unlike antibiotics, the extract does not appear to promote drug-resistance, at least for the 20 generations observed by the researchers.
Eczema is a chronic, relapsing disease characterized by skin that is dry, itchy, inflamed, and prone to infection, including by pathogens such as S. aureus and herpes virus. Although the cause of eczema is unknown, the condition is associated with human genetic mutations, disruption of the skin’s barrier, inflammation-triggering allergens, and imbalances in the skin microbiome.
In 2017, Gallo’s research team discovered that, in healthy human skin, certain strains of Staphylococcus hominis and Staphylococcus epidermis produce potent antimicrobial molecules known as lantibiotics [4]. These beneficial strains are far less common on the skin of people with eczema. The lantibiotics work synergistically with LL-37, an antimicrobial molecule produced by the human immune system, to selectively kill S. aureus, including methicillin-resistant strains (MRSA).
Gallo and his colleagues then examined the safety and therapeutic potential of these beneficial strains isolated from the human skin microbiome. In animal tests, strains of S. hominis and S. epidermis that produce lantibiotics killed S. aureus and blocked production of its toxin.
Gallo’s group has now expanded their work to early studies in humans. In 2021, two independent phase 1 clinical trials [5,6] conducted by Gallo and his colleagues investigated the effects of these strains on people with eczema. These double-blind, placebo-controlled trials involved one-week of topical application of beneficial bacteria to the forearm of adults with S. aureus-positive eczema. The results demonstrated that the treatment was safe, showed a significant decrease in S. aureus, and improved eczema symptoms in most patients. This is encouraging news for those hoping to develop microbiome-targeted therapy for inflammatory skin diseases.
As research on the skin microbiome advances on different fronts, it will provide deeper insight into the multi-faceted microbial communities that are so critical to health and disease. One day, we may even be able to harness the microbiome as a source of therapeutics to alleviate inflammation, promote wound healing, or suppress certain skin cancers.
References:
[1] Integrating cultivation and metagenomics for a multi-kingdom view of skin microbiome diversity and functions. Saheb Kashaf S, Proctor DM, Deming C, Saary P, Hölzer M; NISC Comparative Sequencing Program, Taylor ME, Kong HH, Segre JA, Almeida A, Finn RD. Nat Microbiol. 2022 Jan;7(1):169-179.
[2] Expanded skin virome in DOCK8-deficient patients. Tirosh O, Conlan S, Deming C, Lee-Lin SQ, Huang X; NISC Comparative Sequencing Program, Su HC, Freeman AF, Segre JA, Kong HH. Nat Med. 2018 Dec;24(12):1815-1821.
[3] Identification of a human skin commensal bacterium that selectively kills Cutibacterium acnes. O’Neill AM, Nakatsuji T, Hayachi A, Williams MR, Mills RH, Gonzalez DJ, Gallo RL. J Invest Dermatol. 2020 Aug;140(8):1619-1628.e2.
[4] Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, Shafiq F, Kotol PF, Bouslimani A, Melnik AV, Latif H, Kim JN, Lockhart A, Artis K, David G, Taylor P, Streib J, Dorrestein PC, Grier A, Gill SR, Zengler K, Hata TR, Leung DY, Gallo RL. Sci Transl Med. 2017 Feb 22;9(378):eaah4680.
[5] Development of a human skin commensal microbe for bacteriotherapy of atopic dermatitis and use in a phase 1 randomized clinical trial. Nakatsuji T, Hata TR, Tong Y, Cheng JY, Shafiq F, Butcher AM, Salem SS, Brinton SL, Rudman Spergel AK, Johnson K, Jepson B, Calatroni A, David G, Ramirez-Gama M, Taylor P, Leung DYM, Gallo RL. Nat Med. 2021 Apr;27(4):700-709.
[6] Use of autologous bacteriotherapy to treat Staphylococcus aureus in patients with atopic dermatitis: A randomized double-blind clinical trial. Nakatsuji T, Gallo RL, Shafiq F, Tong Y, Chun K, Butcher AM, Cheng JY, Hata TR. JAMA Dermatol. 2021 Jun 16;157(8):978-82.
Links:
Acne (National Institute of Arthritis and Musculoskeletal and Skin Diseases/NIH)
Atopic Dermatitis (NIAMS)
Cutaneous Microbiome and Inflammation Laboratory, Heidi Kong (NIAMS)
Julie Segre (National Human Genome Research Institute/NIH)
Gallo Lab (University of California, San Diego)
[Note: Acting NIH Director Lawrence Tabak has asked the heads of NIH’s Institutes and Centers (ICs) to contribute occasional guest posts to the blog to highlight some of the cool science that they support and conduct. This is the fifth in the series of NIH IC guest posts that will run until a new permanent NIH director is in place.]
NCI Support for Basic Science Paves Way for Kidney Cancer Drug Belzutifan
Posted on by Norman "Ned" Sharpless, M.D., National Cancer Institute

There’s exciting news for people with von Hippel-Lindau (VHL) disease, a rare genetic disorder that can lead to cancerous and non-cancerous tumors in multiple organs, including the brain, spinal cord, kidney, and pancreas. In August 2021, the U.S. Food and Drug Administration (FDA) approved belzutifan (Welireg), a new drug that has been shown in a clinical trial led by National Cancer Institute (NCI) researchers to shrink some tumors associated with VHL disease [1], which is caused by inherited mutations in the VHL tumor suppressor gene.
As exciting as this news is, relatively few people have this rare disease. The greater public health implication of this advancement is for people with sporadic, or non-inherited, clear cell kidney cancer, which is by far the most common subtype of kidney cancer, with more than 70,000 cases and about 14,000 deaths per year. Most cases of sporadic clear cell kidney cancer are caused by spontaneous mutations in the VHL gene.
This advancement is also a great story of how decades of support for basic science through NCI’s scientists in the NIH Intramural Research Program and its grantees through extramural research funding has led to direct patient benefit. And it’s a reminder that we never know where basic science discoveries might lead.
Belzutifan works by disrupting the process by which the loss of VHL in a tumor turns on a series of molecular processes. These processes involve the hypoxia-inducible factor (HIF) transcription factor and one of its subunits, HIF-2α, that lead to tumor formation.
The unraveling of the complex relationship among VHL, the HIF pathway, and cancer progression began in 1984, when Bert Zbar, Laboratory of Immunobiology, NCI-Frederick; and Marston Linehan, NCI’s Urologic Oncology Branch, set out to find the gene responsible for clear cell kidney cancer. At the time, there were no effective treatments for advanced kidney cancer, and 80 percent of patients died within two years.
Zbar and Linehan started by studying patients with sporadic clear cell kidney cancer, but then turned their focus to investigations of people affected with VHL disease, which predisposes a person to developing clear cell kidney cancer. By studying the patients and the genetic patterns of tumors collected from these patients, the researchers hypothesized that they could find genes responsible for kidney cancer.
Linehan established a clinical program at NIH to study and manage VHL patients, which facilitated the genetic studies. It took nearly a decade, but, in 1993, Linehan, Zbar, and Michael Lerman, NCI-Frederick, identified the VHL gene, which is mutated in people with VHL disease. They soon discovered that tumors from patients with sporadic clear cell kidney cancer also have mutations in this gene.
Subsequently, with NCI support, William G. Kaelin Jr., Dana-Farber Cancer Institute, Boston, discovered that VHL is a tumor suppressor gene that, when inactivated, leads to the accumulation of HIF.
Another NCI grantee, Gregg L. Semenza, Johns Hopkins School of Medicine, Baltimore, identified HIF as a transcription factor. And Peter Ratcliffe, University of Oxford, United Kingdom, discovered that HIF plays a role in blood vessel development and tumor growth.
Kaelin and Ratcliffe simultaneously showed that the VHL protein tags a subunit of HIF for destruction when oxygen levels are high. These results collectively answered a very old question in cell biology: How do cells sense the intracellular level of oxygen?
Subsequent studies by Kaelin, with NCI’s Richard Klausner and Linehan, revealed the critical role of HIF in promoting the growth of clear cell kidney cancer. This work ultimately focused on one member of the HIF family, the HIF-2α subunit, as the key mediator of clear cell kidney cancer growth.
The fundamental work of Kaelin, Semenza, and Ratcliffe earned them the 2019 Nobel Prize in Physiology or Medicine. It also paved the way for drug discovery efforts that target numerous points in the pathway leading to clear cell kidney cancer, including directly targeting the transcriptional activity of HIF-2α with belzutifan.
Clinical trials of belzutifan, including several supported by NCI, demonstrated potent anti-cancer activity in VHL-associated kidney cancer, as well as other VHL-associated tumors, leading to the aforementioned recent FDA approval. This is an important development for patients with VHL disease, providing a first-in-class therapy that is effective and well-tolerated.
We believe this is only the beginning for belzutifan’s use in patients with cancer. A number of trials are now studying the effectiveness of belzutifan for sporadic clear cell kidney cancer. A phase 3 trial is ongoing, for example, to look at the effectiveness of belzutifan in treating people with advanced kidney cancer. And promising results from a phase 2 study show that belzutifan, in combination with cabozantinib, a widely used agent to treat kidney cancer, shrinks tumors in patients previously treated for metastatic clear cell kidney cancer [2].
This is a great scientific story. It shows how studies of familial cancer and basic cell biology lead to effective new therapies that can directly benefit patients. I’m proud that NCI’s support for basic science, both intramurally and extramurally, is making possible many of the discoveries leading to more effective treatments for people with cancer.
References:
[1] Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, Oudard S, Else T, Maranchie JK, Welsh SJ, Thamake S, Park EK, Perini RF, Linehan WM, Srinivasan R; MK-6482-004 Investigators. N Engl J Med. 2021 Nov 25;385(22):2036-2046.
[2] Phase 2 study of the oral hypoxia-inducible factor 2α (HIF-2α) inhibitor MK-6482 in combination with cabozantinib in patients with advanced clear cell renal cell carcinoma (ccRCC). Choueiri TK et al. J Clin Oncol. 2021 Feb 20;39(6_suppl): 272-272.
Links:
Von Hippel-Lindau Disease (Genetic and Rare Diseases Information Center/National Center for Advancing Translational Sciences/NIH)
Clear Cell Renal Cell Carcinoma (National Cancer Institute/NIH)
Belzutifan Approved to Treat Tumors Linked to Inherited Disorder VHL, Cancer Currents Blog, National Cancer Institute, September 21, 2021.
The Long Road to Understanding Kidney Cancer (Intramural Research Program/NIH)
[Note: Acting NIH Director Lawrence Tabak has asked the heads of NIH’s institutes and centers to contribute occasional guest posts to the blog as a way to highlight some of the cool science that they support and conduct. This is the first in the series of NIH institute and center guest posts that will run until a new permanent NIH director is in place.]
Biomedical Research Leads Science’s 2021 Breakthroughs
Posted on by Lawrence Tabak, D.D.S., Ph.D.

Hi everyone, I’m Larry Tabak. I’ve served as NIH’s Principal Deputy Director for over 11 years, and I will be the acting NIH director until a new permanent director is named. In my new role, my day-to-day responsibilities will certainly increase, but I promise to carve out time to blog about some of the latest research progress on COVID-19 and any other areas of science that catch my eye.
I’ve also invited the directors of NIH’s Institutes and Centers (ICs) to join me in the blogosphere and write about some of the cool science in their research portfolios. I will publish a couple of posts to start, then turn the blog over to our first IC director. From there, I envision alternating between posts from me and from various IC directors. That way, we’ll cover a broad array of NIH science and the tremendous opportunities now being pursued in biomedical research.
Since I’m up first, let’s start where the NIH Director’s Blog usually begins each year: by taking a look back at Science’s Breakthroughs of 2021. The breakthroughs were formally announced in December near the height of the holiday bustle. In case you missed the announcement, the biomedical sciences accounted for six of the journal Science’s 10 breakthroughs. Here, I’ll focus on four biomedical breakthroughs, the ones that NIH has played some role in advancing, starting with Science’s editorial and People’s Choice top-prize winner:
Breakthrough of the Year: AI-Powered Predictions of Protein Structure
The biochemist Christian Anfinsen, who had a distinguished career at NIH, shared the 1972 Nobel Prize in Chemistry, for work suggesting that the biochemical interactions among the amino acid building blocks of proteins were responsible for pulling them into the final shapes that are essential to their functions. In his Nobel acceptance speech, Anfinsen also made a bold prediction: one day it would be possible to determine the three-dimensional structure of any protein based on its amino acid sequence alone. Now, with advances in applying artificial intelligence to solve biological problems—Anfinsen’s bold prediction has been realized.
But getting there wasn’t easy. Every two years since 1994, research teams from around the world have gathered to compete against each other in developing computational methods for predicting protein structures from sequences alone. A score of 90 or above means that a predicted structure is extremely close to what’s known from more time-consuming work in the lab. In the early days, teams more often finished under 60.
In 2020, a London-based company called DeepMind made a leap with their entry called AlphaFold. Their deep learning approach—which took advantage of 170,000 proteins with known structures—most often scored above 90, meaning it could solve most protein structures about as well as more time-consuming and costly experimental protein-mapping techniques. (AlphaFold was one of Science’s runner-up breakthroughs last year.)
This year, the NIH-funded lab of David Baker and Minkyung Baek, University of Washington, Seattle, Institute for Protein Design, published that their artificial intelligence approach, dubbed RoseTTAFold, could accurately predict 3D protein structures from amino acid sequences with only a fraction of the computational processing power and time that AlphaFold required [1]. They immediately applied it to solve hundreds of new protein structures, including many poorly known human proteins with important implications for human health.
The DeepMind and RoseTTAFold scientists continue to solve more and more proteins [1,2], both alone and in complex with other proteins. The code is now freely available for use by researchers anywhere in the world. In one timely example, AlphaFold helped to predict the structural changes in spike proteins of SARS-CoV-2 variants Delta and Omicron [3]. This ability to predict protein structures, first envisioned all those years ago, now promises to speed fundamental new discoveries and the development of new ways to treat and prevent any number of diseases, making it this year’s Breakthrough of the Year.
Anti-Viral Pills for COVID-19
The development of the first vaccines to protect against COVID-19 topped Science’s 2020 breakthroughs. This year, we’ve also seen important progress in treating COVID-19, including the development of anti-viral pills.
First, there was the announcement in October of interim data from Merck, Kenilworth, NJ, and Ridgeback Biotherapeutics, Miami, FL, of a significant reduction in hospitalizations for those taking the anti-viral drug molnupiravir [4] (originally developed with an NIH grant to Emory University, Atlanta). Soon after came reports of a Pfizer anti-viral pill that might target SARS-CoV-2, the novel coronavirus that causes COVID-19, even more effectively. Trial results show that, when taken within three days of developing COVID-19 symptoms, the pill reduced the risk of hospitalization or death in adults at high risk of progressing to severe illness by 89 percent [5].
On December 22, the Food and Drug Administration (FDA) granted Emergency Use Authorization (EUA) for Pfizer’s Paxlovid to treat mild-to-moderate COVID-19 in people age 12 and up at high risk for progressing to severe illness, making it the first available pill to treat COVID-19 [6]. The following day, the FDA granted an EUA for Merck’s molnupiravir to treat mild-to-moderate COVID-19 in unvaccinated, high-risk adults for whom other treatment options aren’t accessible or recommended, based on a final analysis showing a 30 percent reduction in hospitalization or death [7].
Additional promising anti-viral pills for COVID-19 are currently in development. For example, a recent NIH-funded preclinical study suggests that a drug related to molnupiravir, known as 4’-fluorouridine, might serve as a broad spectrum anti-viral with potential to treat infections with SARS-CoV-2 as well as respiratory syncytial virus (RSV) [8].
Artificial Antibody Therapies
Before anti-viral pills came on the scene, there’d been progress in treating COVID-19, including the development of monoclonal antibody infusions. Three monoclonal antibodies now have received an EUA for treating mild-to-moderate COVID-19, though not all are effective against the Omicron variant [9]. This is also an area in which NIH’s Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) public-private partnership has made big contributions.
Monoclonal antibodies are artificially produced versions of the most powerful antibodies found in animal or human immune systems, made in large quantities for therapeutic use in the lab. Until recently, this approach had primarily been put to work in the fight against conditions including cancer, asthma, and autoimmune diseases. That changed in 2021 with success using monoclonal antibodies against infections with SARS-CoV-2 as well as respiratory syncytial virus (RSV), human immunodeficiency virus (HIV), and other infectious diseases. This earned them a prominent spot among Science’s breakthroughs of 2021.
Monoclonal antibodies delivered via intravenous infusions continue to play an important role in saving lives during the pandemic. But, there’s still room for improvement, including new formulations highlighted on the blog last year that might be much easier to deliver.
CRISPR Fixes Genes Inside the Body
One of the most promising areas of research in recent years has been gene editing, including CRISPR/Cas9, for fixing misspellings in genes to treat or even cure many conditions. This year has certainly been no exception.
CRISPR is a highly precise gene-editing system that uses guide RNA molecules to direct a scissor-like Cas9 enzyme to just the right spot in the genome to cut out or correct disease-causing misspellings. Science highlights a small study reported in The New England Journal of Medicine by researchers at Intellia Therapeutics, Cambridge, MA, and Regeneron Pharmaceuticals, Tarrytown, NY, in which six people with hereditary transthyretin (TTR) amyloidosis, a condition in which TTR proteins build up and damage the heart and nerves, received an infusion of guide RNA and CRISPR RNA encased in tiny balls of fat [10]. The goal was for the liver to take them up, allowing Cas9 to cut and disable the TTR gene. Four weeks later, blood levels of TTR had dropped by at least half.
In another study not yet published, researchers at Editas Medicine, Cambridge, MA, injected a benign virus carrying a CRISPR gene-editing system into the eyes of six people with an inherited vision disorder called Leber congenital amaurosis 10. The goal was to remove extra DNA responsible for disrupting a critical gene expressed in the eye. A few months later, two of the six patients could sense more light, enabling one of them to navigate a dimly lit obstacle course [11]. This work builds on earlier gene transfer studies begun more than a decade ago at NIH’s National Eye Institute.
Last year, in a research collaboration that included former NIH Director Francis Collins’s lab at the National Human Genome Research Institute (NHGRI), we also saw encouraging early evidence in mice that another type of gene editing, called DNA base editing, might one day correct Hutchinson-Gilford Progeria Syndrome, a rare genetic condition that causes rapid premature aging. Preclinical work has even suggested that gene-editing tools might help deliver long-lasting pain relief. The technology keeps getting better, too. This isn’t the first time that gene-editing advances have landed on Science’s annual Breakthrough of the Year list, and it surely won’t be the last.
The year 2021 was a difficult one as the pandemic continued in the U.S. and across the globe, taking far too many lives far too soon. But through it all, science has been relentless in seeking and finding life-saving answers, from the rapid development of highly effective COVID-19 vaccines to the breakthroughs highlighted above.
As this list also attests, the search for answers has progressed impressively in other research areas during these difficult times. These groundbreaking discoveries are something in which we can all take pride—even as they encourage us to look forward to even bigger breakthroughs in 2022. Happy New Year!
References:
[1] Accurate prediction of protein structures and interactions using a three-track neural network. Baek M, DiMaio F, Anishchenko I, Dauparas J, Grishin NV, Adams PD, Read RJ, Baker D., et al. Science. 2021 Jul 15:eabj8754.
[2] Highly accurate protein structure prediction with AlphaFold. Jumper J, Evans R, Pritzel A, Green T, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. et al. Nature. 2021 Jul 15.
[3] Structural insights of SARS-CoV-2 spike protein from Delta and Omicron variants. Sadek A, Zaha D, Ahmed MS. preprint bioRxiv. 2021 Dec 9.
[5] Pfizer’s novel COVID-19 oral antiviral treatment candidate reduced risk of hospitalization or death by 89% in interim analysis of phase 2/3 EPIC-HR Study. Pfizer. 5 November 52021.
[6] Coronavirus (COVID-19) Update: FDA authorizes first oral antiviral for treatment of COVID-19. Food and Drug Administration. 22 Dec 2021.
[7] Coronavirus (COVID-19) Update: FDA authorizes additional oral antiviral for treatment of COVID-19 in certain adults. Food and Drug Administration. 23 Dec 2021.
[8] 4′-Fluorouridine is an oral antiviral that blocks respiratory syncytial virus and SARS-CoV-2 replication. Sourimant J, Lieber CM, Aggarwal M, Cox RM, Wolf JD, Yoon JJ, Toots M, Ye C, Sticher Z, Kolykhalov AA, Martinez-Sobrido L, Bluemling GR, Natchus MG, Painter GR, Plemper RK. Science. 2021 Dec 2.
[9] Anti-SARS-CoV-2 monoclonal antibodies. NIH COVID-19 Treatment Guidelines. 16 Dec 2021.
[10] CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. Gillmore JD, Gane E, Taubel J, Kao J, Fontana M, Maitland ML, Seitzer J, O’Connell D, Walsh KR, Wood K, Phillips J, Xu Y, Amaral A, Boyd AP, Cehelsky JE, McKee MD, Schiermeier A, Harari O, Murphy A, Kyratsous CA, Zambrowicz B, Soltys R, Gutstein DE, Leonard J, Sepp-Lorenzino L, Lebwohl D. N Engl J Med. 2021 Aug 5;385(6):493-502.
[11] Editas Medicine announces positive initial clinical data from ongoing phase 1/2 BRILLIANCE clinical trial of EDIT-101 For LCA10. Editas Medicine. 29 Sept 2021.
Links:
Structural Biology (National Institute of General Medical Sciences/NIH)
The Structures of Life (NIGMS)
COVID-19 Research (NIH)
2021 Science Breakthrough of the Year (American Association for the Advancement of Science, Washington, D.C)
Encouraging News on Gene Therapy for Hemophilia A
Posted on by Dr. Francis Collins

About 20,000 people in the U.S. live with hemophilia A. It’s a rare X-linked genetic disorder that affects predominantly males and causes their blood to clot poorly when healing wounds. For some, routine daily activities can turn into painful medical emergencies to stop internal bleeding, all because of changes in a single gene that disables an essential clotting protein.
Now, results of an early-stage clinical trial, published recently in the New England Journal of Medicine, demonstrate that gene therapy is within reach to produce the essential clotting factor in people with hemophilia A. The results show that, in most of the 18 adult participants, a refined gene therapy strategy produced lasting expression of factor VIII (FVIII), the missing clotting factor in hemophilia A [1]. In fact, gene therapy helped most participants reduce—or, in some cases, completely eliminate—bleeding events.
Currently, the most-common treatment option for males with hemophilia A is intravenous infusion of FVIII concentrate. Though infused FVIII becomes immediately available in the bloodstream, these treatments aren’t a cure and must be repeated, often weekly or every other day, to prevent or control bleeding.
Gene therapy, however, represents a possible cure for hemophilia A. Earlier clinical trials reported some success using benign adeno-associated viruses (AAVs) as the vector to deliver the therapeutic FVIII gene to cells in the liver, where the clotting protein is made. But after a year, those trial participants had a marked decline in FVIII expression. Follow-up studies then found that the decline continued over time, thought to be at least in part because of an immune response to the AAV vector.
In the new study, an NIH-funded team led by Lindsey George and Katherine High of the Children’s Hospital of Philadelphia and the University of Pennsylvania, tested their refined delivery system. High is also currently with Asklepios BioPharmaceutical, Inc., Chapel Hill, NC. (Back in the 1970s, she and I were medical students in the same class at the University of North Carolina.) The study was also supported by Spark Therapeutics, Philadelphia.
Trial participants received a single infusion of the novel recombinant AAV-based gene therapy called SPK-8011. It is specifically designed to produce FVIII expression in the liver. In this phase 1/2 clinical trial, which evaluates the safety and initial efficacy of a treatment, participants received one of four different doses of SPK-8011. Most also received steroids to prevent or treat the presumed counterproductive immune response to the therapy.
The researchers followed participants for a year after the experimental treatment, and all enrolled in a follow-up trial for continued observation. During this time, researchers detected no major safety concerns, though several patients had increases in blood levels of a liver enzyme.
The great news is all participants produced the missing FVIII after gene therapy. Twelve of the 16 participants were followed for more than two years and had no apparent decrease in clotting factor activity. This is especially noteworthy because it offers the first demonstration of multiyear stable and durable FVIII expression in individuals with hemophilia A following gene transfer.
Even more encouraging, the men in the trial had more than a 92 percent reduction in bleeding episodes on average. Before treatment, most of the men had 8.5 bleeding episodes per year. After treatment, those events dropped to an average of less than one per year. However, two study participants lost FVIII expression within a year of treatment, presumably due to an immune response to the therapeutic AAV. This finding shows that, while steroids help, they don’t always prevent loss of a therapeutic gene’s expression.
Overall, the findings suggest that AAV-based gene therapy can lead to the durable production of FVIII over several years and significantly reduce bleeding events. The researchers are now exploring possibly more effective ways to control the immune response to AAV in expansion of this phase 1/2 investigation before pursuing a larger phase 3 trial. They’re continuing to monitor participants closely to establish safety and efficacy in the months and years to come.
On a related note, the recently announced Bespoke Gene Therapy Consortium (BGTC), a partnership between NIH and industry, will expand the refined gene therapy approach demonstrated here to more rare and ultrarare diseases. That should make these latest findings extremely encouraging news for the millions of people born with other rare genetic conditions caused by known alterations to a single gene.
Reference:
[1] Multiyear Factor VIII expression after AAV Gene transfer for Hemophilia A. George LA, Monahan PE, Eyster ME, Sullivan SK, Ragni MV, Croteau SE, Rasko JEJ, Recht M, Samelson-Jones BJ, MacDougall A, Jaworski K, Noble R, Curran M, Kuranda K, Mingozzi F, Chang T, Reape KZ, Anguela XM, High KA. N Engl J Med. 2021 Nov 18;385(21):1961-1973.
Links:
Hemophilia A (National Center for Advancing Translational Sciences/NIH)
FAQ About Rare Diseases (National Center for Advancing Translational Sciences/NIH)
Bespoke Gene Therapy Consortium (BGTC) (NIH)
Accelerating Medicines Partnership® (AMP®) (NIH)
Lindsey George (University of Pennsylvania, Philadelphia)
Katherine High (University of Pennsylvania)
NIH Support: National Heart, Lung, and Blood Institute
Single-Cell Study Offers New Clue into Causes of Cystic Fibrosis
Posted on by Dr. Francis Collins

More than 30 years ago, I co-led the Michigan-Toronto team that discovered that cystic fibrosis (CF) is caused by an inherited misspelling in the cystic fibrosis transmembrane conductance regulator (CFTR) gene [1]. The CFTR protein’s normal function on the surface of epithelial cells is to serve as a gated channel for chloride ions to pass in and out of the cell. But this function is lost in individuals for whom both copies of CFTR are misspelled. As a consequence, water and salt get out of balance, leading to the production of the thick mucus that leaves people with CF prone to life-threatening lung infections.
It took three decades, but that CFTR gene discovery has now led to the development of a precise triple drug therapy that activates the dysfunctional CFTR protein and provides major benefit to most children and adults with CF. But about 10 percent of individuals with CF have mutations that result in the production of virtually no CFTR protein, which means there is nothing for current triple therapy to correct or activate.
That’s why more basic research is needed to tease out other factors that contribute to CF and, if treatable, could help even more people control the condition and live longer lives with less chronic illness. A recent NIH-supported study, published in the journal Nature Medicine [2], offers an interesting basic clue, and it’s visible in the image above.
The healthy lung tissue (left) shows a well-defined and orderly layer of ciliated cells (green), which use hair-like extensions to clear away mucus and debris. Running closely alongside it is a layer of basal cells (outlined in red), which includes stem cells that are essential for repairing and regenerating upper airway tissue. (DNA indicating the position of cell is stained in blue).
In the CF-affected airways (right), those same cell types are present. However, compared to the healthy lung tissue, they appear to be in a state of disarray. Upon closer inspection, there’s something else that’s unusual if you look carefully: large numbers of a third, transitional cell subtype (outlined in red with green in the nucleus) that combines properties of both basal stem cells and ciliated cells, which is suggestive of cells in transition. The image below more clearly shows these cells (yellow arrows).

The increased number of cells with transitional characteristics suggests an unsuccessful attempt by the lungs to produce more cells capable of clearing the mucus buildup that occurs in airways of people with CF. The data offer an important foundation and reference for continued study.
These findings come from a team led by Kathrin Plath and Brigitte Gomperts, University of California, Los Angeles; John Mahoney, Cystic Fibrosis Foundation, Lexington, MA; and Barry Stripp, Cedars-Sinai, Los Angeles. Together with their lab members, they’re part of a larger research team assembled through the Cystic Fibrosis Foundation’s Epithelial Stem Cell Consortium, which seeks to learn how the disease changes the lung’s cellular makeup and use that new knowledge to make treatment advances.
In this study, researchers analyzed the lungs of 19 people with CF and another 19 individuals with no evidence of lung disease. Those with CF had donated their lungs for research in the process of receiving a lung transplant. Those with healthy lungs were organ donors who died of other causes.
The researchers analyzed, one by one, many thousands of cells from the airway and classified them into subtypes based on their distinctive RNA patterns. Those patterns indicate which genes are switched on or off in each cell, as well as the degree to which they are activated. Using a sophisticated computer-based approach to sift through and compare data, the team created a comprehensive catalog of cell types and subtypes present in healthy airways and in those affected by CF.
The new catalogs also revealed that the airways of people with CF had alterations in the types and proportions of basal cells. Those differences included a relative overabundance of cells that appeared to be transitioning from basal stem cells into the specialized ciliated cells, which are so essential for clearing mucus from the lungs.
We are not yet at our journey’s end when it comes to realizing the full dream of defeating CF. For the 10 percent of CF patients who don’t benefit from the triple-drug therapy, the continuing work to find other treatment strategies should be encouraging news. Keep daring to dream of breathing free. Through continued research, we can make the story of CF into history!
References:
[1] Identification of the cystic fibrosis gene: chromosome walking and jumping. Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, et al. Science.1989 Sep 8;245(4922):1059-65.
[2] Transcriptional analysis of cystic fibrosis airways at single-cell resolution reveals altered epithelial cell states and composition. Carraro G, Langerman J, Sabri S, Lorenzana Z, Purkayastha A, Zhang G, Konda B, Aros CJ, Calvert BA, Szymaniak A, Wilson E, Mulligan M, Bhatt P, Lu J, Vijayaraj P, Yao C, Shia DW, Lund AJ, Israely E, Rickabaugh TM, Ernst J, Mense M, Randell SH, Vladar EK, Ryan AL, Plath K, Mahoney JE, Stripp BR, Gomperts BN. Nat Med. 2021 May;27(5):806-814.
Links:
Cystic Fibrosis (National Heart, Lung, and Blood Institute/NIH)
Kathrin Plath (University of California, Los Angeles)
Brigitte Gomperts (UCLA)
Stripp Lab (Cedars-Sinai, Los Angeles)
Cystic Fibrosis Foundation (Lexington, MA)
Epithelial Stem Cell Consortium (Cystic Fibrosis Foundation, Lexington, MA)
NIH Support: National Heart, Lung, and Blood Institute; National Institute of Diabetes and Digestive and Kidney Diseases; National Institute of General Medical Sciences; National Cancer Institute; National Center for Advancing Translational Sciences
DNA Base Editing May Treat Progeria, Study in Mice Shows
Posted on by Dr. Francis Collins

My good friend Sam Berns was born with a rare genetic condition that causes rapid premature aging. Though Sam passed away in his teens from complications of this condition, called Hutchinson-Gilford progeria syndrome, he’s remembered today for his truly positive outlook on life. Sam expressed it, in part, by his willingness to make adjustments that allowed him, in his words, to put things that he always wanted to do in the “can do” category.
In this same spirit on behalf of the several hundred kids worldwide with progeria and their families, a research collaboration, including my NIH lab, has now achieved a key technical advance to move non-heritable gene editing another step closer to the “can do” category to treat progeria. As published in the journal Nature, our team took advantage of new gene-editing tools to correct for the first time a single genetic misspelling responsible for progeria in a mouse model, with dramatically beneficial effects [1, 2]. This work also has implications for correcting similar single-base typos that cause other inherited genetic disorders.
The outcome of this work is incredibly gratifying for me. In 2003, my NIH lab discovered the DNA mutation that causes progeria. One seemingly small glitch—swapping a “T” in place of a “C” in a gene called lamin A (LMNA)—leads to the production of a toxic protein now known as progerin. Without treatment, children with progeria develop normally intellectually but age at an exceedingly rapid pace, usually dying prematurely from heart attacks or strokes in their early teens.
The discovery raised the possibility that correcting this single-letter typo might one day help or even cure children with progeria. But back then, we lacked the needed tools to edit DNA safely and precisely. To be honest, I didn’t think that would be possible in my lifetime. Now, thanks to advances in basic genomic research, including work that led to the 2020 Nobel Prize in Chemistry, that’s changed. In fact, there’s been substantial progress toward using gene-editing technologies, such as the CRISPR editing system, for treating or even curing a wide range of devastating genetic conditions, such as sickle cell disease and muscular dystrophy
It turns out that the original CRISPR system, as powerful as it is, works better at knocking out genes than correcting them. That’s what makes some more recently developed DNA editing agents and approaches so important. One of them, which was developed by David R. Liu, Broad Institute of MIT and Harvard, Cambridge, MA, and his lab members, is key to these latest findings on progeria, reported by a team including my lab in NIH’s National Human Genome Research Institute and Jonathan Brown, Vanderbilt University Medical Center, Nashville, TN.
The relatively new gene-editing system moves beyond knock-outs to knock-ins [3,4]. Here’s how it works: Instead of cutting DNA as CRISPR does, base editors directly convert one DNA letter to another by enzymatically changing one DNA base to become a different base. The result is much like the find-and-replace function used to fix a typo in a word processor. What’s more, the gene editor does this without cutting the DNA.
Our three labs (Liu, Brown, and Collins) first teamed up with the Progeria Research Foundation, Peabody, MA, to obtain skin cells from kids with progeria. In lab studies, we found that base editors, targeted by an appropriate RNA guide, could successfully correct the LMNA gene in those connective tissue cells. The treatment converted the mutation back to the normal gene sequence in an impressive 90 percent of the cells.
But would it work in a living animal? To get the answer, we delivered a single injection of the DNA-editing apparatus into nearly a dozen mice either three or 14 days after birth, which corresponds in maturation level roughly to a 1-year-old or 5-year-old human. To ensure the findings in mice would be as relevant as possible to a future treatment for use in humans, we took advantage of a mouse model of progeria developed in my NIH lab in which the mice carry two copies of the human LMNA gene variant that causes the condition. Those mice develop nearly all of the features of the human illness
In the live mice, the base-editing treatment successfully edited in the gene’s healthy DNA sequence in 20 to 60 percent of cells across many organs. Many cell types maintained the corrected DNA sequence for at least six months—in fact, the most vulnerable cells in large arteries actually showed an almost 100 percent correction at 6 months, apparently because the corrected cells had compensated for the uncorrected cells that had died out. What’s more, the lifespan of the treated animals increased from seven to almost 18 months. In healthy mice, that’s approximately the beginning of old age.
This is the second notable advance in therapeutics for progeria in just three months. Last November, based on preclinical work from my lab and clinical trials conducted by the Progeria Research Foundation in Boston, the Food and Drug Administration (FDA) approved the first treatment for the condition. It is a drug called Zokinvy, and works by reducing the accumulation of progerin [5]. With long-term treatment, the drug is capable of extending the life of kids with progeria by 2.5 years and sometimes more. But it is not a cure.
We are hopeful this gene editing work might eventually lead to a cure for progeria. But mice certainly aren’t humans, and there are still important steps that need to be completed before such a gene-editing treatment could be tried safely in people. In the meantime, base editors and other gene editing approaches keep getting better—with potential application to thousands of genetic diseases where we know the exact gene misspelling. As we look ahead to 2021, the dream envisioned all those years ago about fixing the tiny DNA typo responsible for progeria is now within our grasp and getting closer to landing in the “can do” category.
References:
[1] In vivo base editing rescues Hutchinson-Gilford Progeria Syndrome in mice. Koblan LW et al. Nature. 2021 Jan 6.
[2] Base editor repairs mutation found in the premature-ageing syndrome progeria. Vermeij WP, Hoeijmakers JHJ. Nature. 6 Jan 2021.
[3] Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Nature. 2016 May 19;533(7603):420-424.
[4] Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR. Nature. 2017 Nov 23;551(7681):464-471.
[5] FDA approves first treatment for Hutchinson-Gilford progeria syndrome and some progeroid laminopathies. Food and Drug Administration. 2020 Nov 20.
Links:
Progeria (Genetic and Rare Diseases Information Center/NIH)
What are Genome Editing and CRISPR-Cas9? (National Library of Medicine/NIH)
Somatic Cell Genome Editing Program (Common Fund/NIH)
David R. Liu (Harvard University, Cambridge, MA)
Collins Group (National Human Genome Research Institute/NIH)
Jonathan Brown (Vanderbilt University Medical Center, Nashville, TN)
NIH Support: National Human Genome Research Institute; National Center for Advancing Translational Sciences; National Institute of Biomedical Imaging and Bioengineering; National Institute of Allergy and Infectious Diseases; National Institute of General Medical Sciences; Common Fund
Encouraging News for Kids with Neurofibromatosis Type 1
Posted on by Dr. Francis Collins
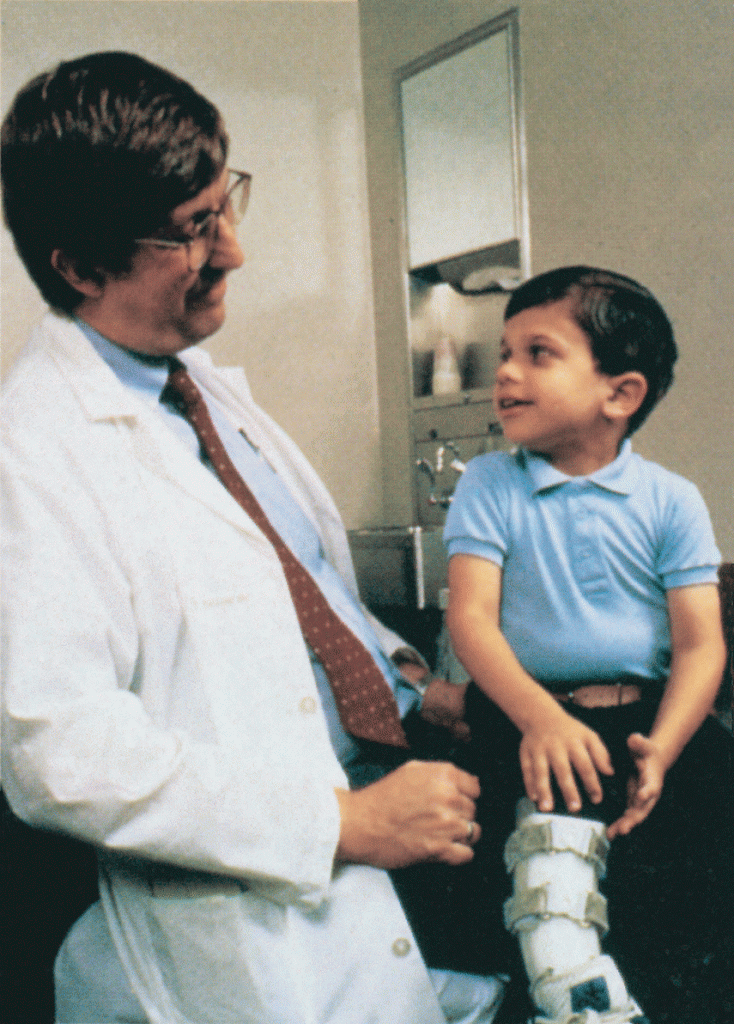
Amid all the headlines and uncertainty surrounding the current COVID-19 pandemic, it’s easy to overlook the important progress that biomedical research is making against other diseases. So, today, I’m pleased to share word of what promises to be the first effective treatment to help young people suffering from the consequences of a painful, often debilitating genetic disorder called neurofibromatosis type 1 (NF1).
This news is particularly meaningful to me because, 30 years ago, I led a team that discovered the gene that underlies NF1. About 1 in 3,000 babies are born with NF1. In about half of those affected, a type of tumor called a plexiform neurofibroma arises along nerves in the skin, face, and other parts of the body. While plexiform neurofibromas are not cancerous, they grow steadily and can lead to severe pain and a range of other health problems, including vision and hearing loss, hypertension, and mobility issues.
The good news is the results of a phase II clinical trial involving NF1, just published in the New England Journal of Medicine. The trial was led by Brigitte Widemann and Andrea Gross, researchers in the Center for Cancer Research at NIH’s National Cancer Institute.
The trial’s results confirm that a drug originally developed to treat cancer, called selumetinib, can shrink inoperable tumors in many children with NF1. They also establish that the drug can help affected kids make significant improvements in strength, range of motion, and quality of life. While selumetinib is not a cure, and further studies are still needed to see how well the treatment works in the long term, these results suggest that the first effective treatment for NF1 is at last within our reach.
Selumetinib blocks a protein in human cells called MEK. This protein is involved in a major cellular pathway known as RAS that can become dysregulated and give rise to various cancers. By blocking the MEK protein in animal studies and putting the brakes on the RAS pathway when it malfunctions, selumetinib showed great initial promise as a cancer drug.
Selumetinib was first tested several years ago in people with a variety of other cancers, including ovarian and non-small cell lung cancers. The clinical research looked good at first but eventually stalled, and so did much of the initial enthusiasm for selumetinib.
But the enthusiasm picked up when researchers considered repurposing the drug to treat NF1. The neurofibromas associated with the condition were known to arise from a RAS-activating loss of the NF1 gene. It made sense that blocking the MEK protein might blunt the overactive RAS signal and help to shrink these often-inoperable tumors.
An earlier phase 1 safety trial looked promising, showing for the first time that the drug could, in some cases, shrink large NF1 tumors [2]. This fueled further research, and the latest study now adds significantly to that evidence.
In the study, Widemann and colleagues enrolled 50 children with NF1, ranging in age from 3 to 17. Their tumor-related symptoms greatly affected their wellbeing and ability to thrive, including disfigurement, limited strength and motion, and pain. Children received selumetinib alone orally twice a day and went in for assessments at least every four months.
As of March 2019, 35 of the 50 children in the ongoing study had a confirmed partial response, meaning that their tumors had shrunk by more than 20 percent. Most had maintained that response for a year or more. More importantly, the kids also felt less pain and were more able to enjoy life.
It’s important to note that the treatment didn’t work for everyone. Five children stopped taking the drug due to side effects. Six others progressed while on the drug, though five of them had to reduce their dose because of side effects before progressing. Nevertheless, for kids with NF1 and their families, this is a big step forward.
Drug developer AstraZeneca, working together with the researchers, has submitted a New Drug Application to the Food and Drug Administration (FDA). While they’re eagerly awaiting the FDA’s decision, the work continues.
The researchers want to learn much more about how the drug affects the health and wellbeing of kids who take it over the long term. They’re also curious whether it could help to prevent the growth of large tumors in kids who begin taking it earlier in the course of the disease, and whether it might benefit other features of the disorder. They will continue to look ahead to other potentially promising treatments or treatment combinations that may further help, and perhaps one day even cure, kids with NF1. So, even while we cope with the COVID-19 pandemic, there are reasons to feel encouraged and grateful for continued progress made throughout biomedical research.
References:
[1] Selumitinib in children with inoperable plexiform neurofibromas. New England Journal of Medicine. Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, Weiss B, Kim A, Bornhorst M, Shah AC, Martin S, Roderick MC, Pichard DC, Carbonell A, Paul SM, Therrien J, Kapustina O, Heisey K, Clapp DW, Zhang C, Peer CJ, Figg WD, Smith M, Glod J, Blakeley JO, Steinberg SM, Venzon DJ, Doyle LA, Widemann BC. 18 March 2020. N Engl J Med. 2020 Mar 18. [Epub ahead of publication.]
[2] Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, Whitcomb P, Martin S, Aschbacher-Smith LE, Rizvi TA, Wu J, Ershler R, Wolters P1, Therrien J, Glod J, Belasco JB, Schorry E, Brofferio A, Starosta AJ, Gillespie A, Doyle AL, Ratner N, Widemann BC. N Engl J Med. 2016 Dec 29;375(26):2550-2560.
Links:
Neurofibromatosis Fact Sheet (National Institute of Neurological Disorders and Stroke/NIH)
Brigitte Widemann (National Cancer Institute/NIH)
Andrea Gross (National Cancer Institute/NIH)
NIH Support: National Cancer Institute
Seven Questions for a Rare Disease Warrior
Posted on by Dr. Francis Collins

Credit: National Disease Research Interchange, Philadelphia
Tomorrow is Rare Disease Day at NIH, marking the 12th year that this annual event has been held on the NIH campus. Similar gatherings have been organized independently around the world this week, all to raise awareness for the nearly 7,000 rare diseases, some affecting just a few dozen people. But, collectively, rare diseases are hardly rare. One in 10 Americans has a rare disease (defined as affecting 200,000 or fewer individuals in the US), and about half are children. Without needed treatments, about 30 percent of these children will die by age 5.
To join everyone in raising awareness, I wanted to feature on my blog a unique perspective about rare diseases, and David Fajgenbaum certainly has one. Fajgenbaum is an immunologist and NIH grantee at the Perelman School of Medicine, University of Pennsylvania, Philadelphia. When Fajgenbaum isn’t running studies or clinical trials, he must remain vigilant of his own health. Fajgenbaum has a rare disease called idiopathic multicentric Castleman disease (iMCD), and this devastating condition, which emerged while he was in medical school, nearly claimed his life several times.
Now 34 years old and in a long remission, Fajgenbaum can discuss rare diseases as a doctor, as a patient, as a researcher, and as an advocate. His personal journey, published in his recent book Chasing My Cure, is a gripping read. Fajgenbaum was kind enough to answer a few of my questions on rare diseases and share some of his lessons learned.
The last time that I saw you, David, you looked great. How long have you been in remission?
I have been in remission for 73.83 months. I say 73.83, because I know that I can’t round up—I may relapse tomorrow. But I also refuse to round down because so many colleagues and I have worked so hard for every day of remission for me and other patients with my disease.
For me, every day is particularly special, because I never thought that I would be alive this long. As you know, I became deathly ill during medical school in 2010 and even had my last rites read to me when my doctors didn’t think I would survive. I was eventually diagnosed with idiopathic multicentric Castleman disease (iMCD), which is like a deadly cross between cancer and autoimmunity. Chemotherapy saved my life, but I would go on to have four near-death relapses.
After one of those relapses, I got out of the hospital and dedicated my life to conducting iMCD research and co-founded the Castleman Disease Collaborative Network (CDCN). Later, I identified a particular cellular pathway called mTOR that was highly active in my samples. I began testing on myself an mTOR inhibitor [sirolimus]—developed 30 years before and approved for kidney transplantation but never considered for iMCD. It’s this drug that has kept me in remission for the last 73.83 months and helped other people. During this time, I’ve been able to marry my wife, have a daughter, help launch a new center at Penn specializing in rare diseases, and write a book to share my personal journey with others.
As a physician-scientist and as a person with a rare disease, what have you learned about the biomedical research process?
I’ve learned so much, but I’d like to highlight three lessons in particular. First, we must leverage all perspectives to prioritize research and give us the best chance of translating research into meaningful breakthroughs. The traditional approach to rare disease research involves a subset of researchers within a rare disease field submitting their best ideas for funding and a panel selecting the best applicant.
Through the CDCN, we’ve spearheaded a new approach called the Collaborative Network Approach, where we crowdsource research questions from the entire community of patients, physicians, and researchers (not just a subset of researchers) and then recruit the best researchers in the world (not just from within the Castleman disease field) to perform the prioritized studies. We’re now working to improve and spread this approach to other diseases.
Second, collaboration between all players is critical. Patient advocacy groups are uniquely positioned to serve as the glue between all stakeholders. Researchers and physicians need to share ideas, data, and samples with one another. Patients need to be actively involved in research question prioritization and study design. Biopharma and the Food and Drug Administration (FDA) need to be engaged early in the process of research discoveries and drug development.
Third, we must leverage all 1,500-plus, existing FDA-approved drugs to help as many patients without any options as quickly as possible. As you know, less than 5 percent of the nearly 7,000 rare diseases have an FDA-approved therapy, but many diseases share similar cellular and genetic defects that could make them susceptible to the same drugs. I’m literally alive today thanks to a drug developed for another disease. How many of the drugs approved for one disease may be effective for many of the 7,000 diseases without any? I don’t know the answer, but I hope we can begin to address this important question and incentivize repurposing.
In your experience, how can people with rare diseases help to advance progress for their conditions?
There is so much work to be done for so many rare diseases. Sometimes it can feel so overwhelming and like “what can I really do?”
But I’ve learned that there are so many ways that we can each contribute and so many incredible examples of advocates who have made a difference for themselves and those that they love. Cystic fibrosis and chordoma are just two of many examples where patient-advocates have been critical partners in transforming their diseases.
People with rare diseases can raise funds for research. Every dollar truly counts. We can work with existing organizations for our disease to ensure that those funds are distributed as efficiently and effectively as possible. If there are major gaps within our rare disease fields that aren’t being addressed by existing organizations, we can start new rare disease organizations (but we should try to avoid this whenever possible). We can contribute samples and data towards research, participate in clinical trials, and share with other patients about our experiences. We can advocate for new drug development and repurposing already-FDA approved drugs for our diseases.
What would you tell other researchers who are studying rare diseases?
I would tell other rare disease researchers that you are doing such important work. You give us hope that a treatment can be identified that will change our lives. It’s an incredible responsibility and incredibly stressful. There are unfortunately far too many scientific questions and diseases with major unmet need for any of us to compete over the use of samples and data. We have to share these within our fields. And we must also work together across rare diseases. We can’t continue to reinvent the wheel; we must share learnings with one another
I enjoyed doing the CastleMan Warrior Flex with you. Tell us more about what it represents?
Doing the CastleMan Warrior Flex with you is one of my favorite pictures. In fact, it’s hanging up in my office.
Castleman disease was named after Dr. Benjamin Castleman, who first described our disease in 1954. We have repurposed the “Castleman” name to be a “CastleMan Warrior” (below is our cartoon mascot). We do the “CastleMan Warrior” Flex to raise awareness for Castleman disease and rare diseases generally—we’re all warriors in the rare disease space.

What are your future plans as a rare disease advocate and as a researcher?
We’ve made a lot of progress for Castleman disease: we’ve advanced our findings about mTOR towards a clinical trial, gained approval for the treatment siltuximab for iMCD, developed diagnostic criteria and treatment guidelines, and invested about $1.5 million into Castleman disease research, which has led to over $7 million in additional funding from other sources.
But we still have important work ahead of us. The treatments sirolimus and siltuximab work for only a portion of all iMCD patients. We need to identify more effective treatments for all forms of Castleman disease.
I will continue to study Castleman disease and other diseases at the intersection of autoimmunity and oncology to gain insights into how the immune system works in myriad diseases. In parallel, I will continue to advocate for the adoption of the “Collaborative Network Approach” to crowdsource all stakeholder perspectives as well as for new models for drug repurposing.
Any other issues that you’d like to address?
I feel a responsibility to share with the world the lessons that I’ve learned about life from nearly dying five times. This is a major reason that I wrote my book.
One lesson that I think about a lot is related to my growing up playing football. Some of my games were extended into an overtime period to decide the outcome. In overtime, every second counts and you’re totally focused on what’s important. I’ve lived with that exact same feeling ever since I had my last rites read to me.
I’ve also learned that humor can be incredibly powerful. You may think that a good laugh may be the last thing that you’d want to do when you’re dying in the ICU. But laughing with the people that I love actually helped me feel like I could transcend my illness, and it helped to connect us.
My greatest regrets on my deathbed were not things that I had done or said. I regretted what I didn’t do or didn’t say and that I would no longer be able to do. I now follow the motto: “Think It, Do It.” In other words, we should reflect on what we’re hoping for and then turn our hopes into action.
Finally, I’ve learned that it really takes a strong team to make a difference in the world, especially against diseases. If it was just me on my own, we would have made less than 1 percent of the progress that’s been achieved. I hope that all rare disease warriors will join together into strong teams, armies even, and make a difference in the world.
Links:
Multicentric Castleman Disease (Genetic and Rare Diseases Information Center/NIH)
Castleman Disease Collaborative Network (Paso Robles, CA)
His Doctors Were Stumped. Then He Took Over (New York Times, February 4, 2017)
Video: Chasing My Cure: Dr. David Fajgenbaum’s Lessons From His Rare Disease And On Finding Cures For Others (Exponential Medicine, November 4, 2019)
Rare Disease Day at NIH 2020 (National Center for Advancing Translational Sciences/NIH)
Tackling Fibrosis with Synthetic Materials
Posted on by Dr. Francis Collins

When injury strikes a limb or an organ, our bodies usually heal quickly and correctly. But for some people, the healing process doesn’t shut down properly, leading to excess fibrous tissue, scarring, and potentially life-threatening organ damage.
This permanent scarring, known as fibrosis, can occur in almost every tissue of the body, including the heart and lungs. With support from a 2019 NIH Director’s New Innovator Award, April Kloxin is applying her expertise in materials science and bioengineering to build sophisticated fibrosis-in-a-dish models for unraveling this complex process in her lab at the University of Delaware, Newark.
Though Kloxin is interested in all forms of fibrosis, she’s focusing first on the incurable and often-fatal lung condition called idiopathic pulmonary fibrosis (IPF). This condition, characterized by largely unexplained thickening and stiffening of lung tissue, is diagnosed in about 50,000 people each year in the United States.
IPF remains poorly understood, in part because it often is diagnosed when the disease is already well advanced. Kloxin hopes to turn back the clock and start to understand the disease at an earlier stage, when interventions might be more successful. The key is to develop a model that better recapitulates the complexity and irreversibility of the disease process in people.
Building that better model starts with simulating the meshwork of collagen and other proteins in the extracellular matrix (ECM) that undergird every tissue and organ in the body. The ECM’s interactions with our cells are essential in wound healing and, when things go wrong, also in causing fibrosis.
Kloxin will build three-dimensional hydrogels, crosslinked sponge-like networks of polymers, peptides, and proteins, with structures that more accurately capture the biological complexities of human tissues, including the ECMs within fibrous collagen-rich microenvironments. Her synthetic matrices can be triggered with light to lock in place and stiffen. The matrices also will make it possible to culture the lung’s epithelium, or outermost layer of cells, and connective tissue that surrounds it, to study cellular responses as the model shifts from a healthy and flexible to a stiffened, disease-like state.
Kloxin and her team will also integrate into their model system lung cells that have been engineered to fluoresce or light up under a microscope when the wound-healing program activates. Such fluorescent reporters will allow her team to watch for the first time how different cells and their nearby microenvironment respond as the composition of the ECM changes and stiffens. With this system, she’ll also be able to search for small molecules with the ability to turn off excessive wound healing.
The hope is that what’s learned with her New Innovator Award will lead to fresh insights and ultimately new treatments for this mysterious, hard-to-treat condition. But the benefits could be even more wide-ranging. Kloxin thinks that her findings will have implications for the prevention and treatment of other fibrotic diseases as well.
Links:
Idiopathic Pulmonary Fibrosis (National Heart, Lung, and Blood Institute/NIH)
April Kloxin Group (University of Delaware, Newark)
Kloxin Project Information (NIH RePORTER)
NIH Director’s New Innovator Award (Common Fund)
NIH Support: Common Fund; National Heart, Lung, and Blood Institute
Celebrating 2019 Biomedical Breakthroughs
Posted on by Dr. Francis Collins

Happy New Year! As we say goodbye to the Teens, let’s take a look back at 2019 and some of the groundbreaking scientific discoveries that closed out this remarkable decade.
Each December, the reporters and editors at the journal Science select their breakthrough of the year, and the choice for 2019 is nothing less than spectacular: An international network of radio astronomers published the first image of a black hole, the long-theorized cosmic singularity where gravity is so strong that even light cannot escape [1]. This one resides in a galaxy 53 million light-years from Earth! (A light-year equals about 6 trillion miles.)
Though the competition was certainly stiff in 2019, the biomedical sciences were well represented among Science’s “runner-up” breakthroughs. They include three breakthroughs that have received NIH support. Let’s take a look at them:
In a first, drug treats most cases of cystic fibrosis: Last October, two international research teams reported the results from phase 3 clinical trials of the triple drug therapy Trikafta to treat cystic fibrosis (CF). Their data showed Trikafta effectively compensates for the effects of a mutation carried by about 90 percent of people born with CF. Upon reviewing these impressive data, the Food and Drug Administration (FDA) approved Trikafta, developed by Vertex Pharmaceuticals.
The approval of Trikafta was a wonderful day for me personally, having co-led the team that isolated the CF gene 30 years ago. A few years later, I wrote a song called “Dare to Dream” imagining that wonderful day when “the story of CF is history.” Though we’ve still got more work to do, we’re getting a lot closer to making that dream come true. Indeed, with the approval of Trikafta, most people with CF have for the first time ever a real chance at managing this genetic disease as a chronic condition over the course of their lives. That’s a tremendous accomplishment considering that few with CF lived beyond their teens as recently as the 1980s.
Such progress has been made possible by decades of work involving a vast number of researchers, many funded by NIH, as well as by more than two decades of visionary and collaborative efforts between the Cystic Fibrosis Foundation and Aurora Biosciences (now, Vertex) that built upon that fundamental knowledge of the responsible gene and its protein product. Not only did this innovative approach serve to accelerate the development of therapies for CF, it established a model that may inform efforts to develop therapies for other rare genetic diseases.
Hope for Ebola patients, at last: It was just six years ago that news of a major Ebola outbreak in West Africa sounded a global health emergency of the highest order. Ebola virus disease was then recognized as an untreatable, rapidly fatal illness for the majority of those who contracted it. Though international control efforts ultimately contained the spread of the virus in West Africa within about two years, over 28,600 cases had been confirmed leading to more than 11,000 deaths—marking the largest known Ebola outbreak in human history. Most recently, another major outbreak continues to wreak havoc in northeastern Democratic Republic of Congo (DRC), where violent civil unrest is greatly challenging public health control efforts.
As troubling as this news remains, 2019 brought a needed breakthrough for the millions of people living in areas susceptible to Ebola outbreaks. A randomized clinical trial in the DRC evaluated four different drugs for treating acutely infected individuals, including an antibody against the virus called mAb114, and a cocktail of anti-Ebola antibodies referred to as REGN-EB3. The trial’s preliminary data showed that about 70 percent of the patients who received either mAb114 or the REGN-EB3 antibody cocktail survived, compared with about half of those given either of the other two medicines.
So compelling were these preliminary results that the trial, co-sponsored by NIH’s National Institute of Allergy and Infectious Diseases (NIAID) and the DRC’s National Institute for Biomedical Research, was halted last August. The results were also promptly made public to help save lives and stem the latest outbreak. All Ebola patients in the DRC treatment centers now are treated with one or the other of these two options. The trial results were recently published.
The NIH-developed mAb114 antibody and the REGN-EB3 cocktail are the first therapeutics to be shown in a scientifically rigorous study to be effective at treating Ebola. This work also demonstrates that ethically sound clinical research can be conducted under difficult conditions in the midst of a disease outbreak. In fact, the halted study was named Pamoja Tulinde Maisha (PALM), which means “together save lives” in Kiswahili.
To top off the life-saving progress in 2019, the FDA just approved the first vaccine for Ebola. Called Ervebo (earlier rVSV-ZEBOV), this single-dose injectable vaccine is a non-infectious version of an animal virus that has been genetically engineered to carry a segment of a gene from the Zaire species of the Ebola virus—the virus responsible for the current DRC outbreak and the West Africa outbreak. Because the vaccine does not contain the whole Zaire virus, it can’t cause Ebola. Results from a large study in Guinea conducted by the WHO indicated that the vaccine offered substantial protection against Ebola virus disease. Ervebo, produced by Merck, has already been given to over 259,000 individuals as part of the response to the DRC outbreak. The NIH has supported numerous clinical trials of the vaccine, including an ongoing study in West Africa.
Microbes combat malnourishment: Researchers discovered a few years ago that abnormal microbial communities, or microbiomes, in the intestine appear to contribute to childhood malnutrition. An NIH-supported research team followed up on this lead with a study of kids in Bangladesh, and it published last July its groundbreaking finding: that foods formulated to repair the “gut microbiome” helped malnourished kids rebuild their health. The researchers were able to identify a network of 15 bacterial species that consistently interact in the gut microbiomes of Bangladeshi children. In this month-long study, this bacterial network helped the researchers characterize a child’s microbiome and/or its relative state of repair.
But a month isn’t long enough to determine how the new foods would help children grow and recover. The researchers are conducting a similar study that is much longer and larger. Globally, malnutrition affects an estimated 238 million children under the age 5, stunting their normal growth, compromising their health, and limiting their mental development. The hope is that these new foods and others adapted for use around the world soon will help many more kids grow up to be healthy adults.
Measles Resurgent: The staff at Science also listed their less-encouraging 2019 Breakdowns of the Year, and unfortunately the biomedical sciences made the cut with the return of measles in the U.S. Prior to 1963, when the measles vaccine was developed, 3 to 4 million Americans were sickened by measles each year. Each year about 500 children would die from measles, and many more would suffer lifelong complications. As more people were vaccinated, the incidence of measles plummeted. By the year 2000, the disease was even declared eliminated from the U.S.
But, as more parents have chosen not to vaccinate their children, driven by the now debunked claim that vaccines are connected to autism, measles has made a very preventable comeback. Last October, the Centers for Disease Control and Prevention (CDC) reported an estimated 1,250 measles cases in the United States at that point in 2019, surpassing the total number of cases reported annually in each of the past 25 years.
The good news is those numbers can be reduced if more people get the vaccine, which has been shown repeatedly in many large and rigorous studies to be safe and effective. The CDC recommends that children should receive their first dose by 12 to 15 months of age and a second dose between the ages of 4 and 6. Older people who’ve been vaccinated or have had the measles previously should consider being re-vaccinated, especially if they live in places with low vaccination rates or will be traveling to countries where measles are endemic.
Despite this public health breakdown, 2019 closed out a memorable decade of scientific discovery. The Twenties will build on discoveries made during the Teens and bring us even closer to an era of precision medicine to improve the lives of millions of Americans. So, onward to 2020—and happy New Year!
Reference:
[1] 2019 Breakthrough of the Year. Science, December 19, 2019.
NIH Support: These breakthroughs represent the culmination of years of research involving many investigators and the support of multiple NIH institutes.
Next Page