cystic fibrosis
Single-Cell Study Offers New Clue into Causes of Cystic Fibrosis
Posted on by Dr. Francis Collins

More than 30 years ago, I co-led the Michigan-Toronto team that discovered that cystic fibrosis (CF) is caused by an inherited misspelling in the cystic fibrosis transmembrane conductance regulator (CFTR) gene [1]. The CFTR protein’s normal function on the surface of epithelial cells is to serve as a gated channel for chloride ions to pass in and out of the cell. But this function is lost in individuals for whom both copies of CFTR are misspelled. As a consequence, water and salt get out of balance, leading to the production of the thick mucus that leaves people with CF prone to life-threatening lung infections.
It took three decades, but that CFTR gene discovery has now led to the development of a precise triple drug therapy that activates the dysfunctional CFTR protein and provides major benefit to most children and adults with CF. But about 10 percent of individuals with CF have mutations that result in the production of virtually no CFTR protein, which means there is nothing for current triple therapy to correct or activate.
That’s why more basic research is needed to tease out other factors that contribute to CF and, if treatable, could help even more people control the condition and live longer lives with less chronic illness. A recent NIH-supported study, published in the journal Nature Medicine [2], offers an interesting basic clue, and it’s visible in the image above.
The healthy lung tissue (left) shows a well-defined and orderly layer of ciliated cells (green), which use hair-like extensions to clear away mucus and debris. Running closely alongside it is a layer of basal cells (outlined in red), which includes stem cells that are essential for repairing and regenerating upper airway tissue. (DNA indicating the position of cell is stained in blue).
In the CF-affected airways (right), those same cell types are present. However, compared to the healthy lung tissue, they appear to be in a state of disarray. Upon closer inspection, there’s something else that’s unusual if you look carefully: large numbers of a third, transitional cell subtype (outlined in red with green in the nucleus) that combines properties of both basal stem cells and ciliated cells, which is suggestive of cells in transition. The image below more clearly shows these cells (yellow arrows).

The increased number of cells with transitional characteristics suggests an unsuccessful attempt by the lungs to produce more cells capable of clearing the mucus buildup that occurs in airways of people with CF. The data offer an important foundation and reference for continued study.
These findings come from a team led by Kathrin Plath and Brigitte Gomperts, University of California, Los Angeles; John Mahoney, Cystic Fibrosis Foundation, Lexington, MA; and Barry Stripp, Cedars-Sinai, Los Angeles. Together with their lab members, they’re part of a larger research team assembled through the Cystic Fibrosis Foundation’s Epithelial Stem Cell Consortium, which seeks to learn how the disease changes the lung’s cellular makeup and use that new knowledge to make treatment advances.
In this study, researchers analyzed the lungs of 19 people with CF and another 19 individuals with no evidence of lung disease. Those with CF had donated their lungs for research in the process of receiving a lung transplant. Those with healthy lungs were organ donors who died of other causes.
The researchers analyzed, one by one, many thousands of cells from the airway and classified them into subtypes based on their distinctive RNA patterns. Those patterns indicate which genes are switched on or off in each cell, as well as the degree to which they are activated. Using a sophisticated computer-based approach to sift through and compare data, the team created a comprehensive catalog of cell types and subtypes present in healthy airways and in those affected by CF.
The new catalogs also revealed that the airways of people with CF had alterations in the types and proportions of basal cells. Those differences included a relative overabundance of cells that appeared to be transitioning from basal stem cells into the specialized ciliated cells, which are so essential for clearing mucus from the lungs.
We are not yet at our journey’s end when it comes to realizing the full dream of defeating CF. For the 10 percent of CF patients who don’t benefit from the triple-drug therapy, the continuing work to find other treatment strategies should be encouraging news. Keep daring to dream of breathing free. Through continued research, we can make the story of CF into history!
References:
[1] Identification of the cystic fibrosis gene: chromosome walking and jumping. Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, et al. Science.1989 Sep 8;245(4922):1059-65.
[2] Transcriptional analysis of cystic fibrosis airways at single-cell resolution reveals altered epithelial cell states and composition. Carraro G, Langerman J, Sabri S, Lorenzana Z, Purkayastha A, Zhang G, Konda B, Aros CJ, Calvert BA, Szymaniak A, Wilson E, Mulligan M, Bhatt P, Lu J, Vijayaraj P, Yao C, Shia DW, Lund AJ, Israely E, Rickabaugh TM, Ernst J, Mense M, Randell SH, Vladar EK, Ryan AL, Plath K, Mahoney JE, Stripp BR, Gomperts BN. Nat Med. 2021 May;27(5):806-814.
Links:
Cystic Fibrosis (National Heart, Lung, and Blood Institute/NIH)
Kathrin Plath (University of California, Los Angeles)
Brigitte Gomperts (UCLA)
Stripp Lab (Cedars-Sinai, Los Angeles)
Cystic Fibrosis Foundation (Lexington, MA)
Epithelial Stem Cell Consortium (Cystic Fibrosis Foundation, Lexington, MA)
NIH Support: National Heart, Lung, and Blood Institute; National Institute of Diabetes and Digestive and Kidney Diseases; National Institute of General Medical Sciences; National Cancer Institute; National Center for Advancing Translational Sciences
Antibody Response Affects COVID-19 Outcomes in Kids and Adults
Posted on by Dr. Francis Collins

Doctors can’t reliably predict whether an adult newly diagnosed with COVID-19 will recover quickly or battle life-threatening complications. The same is true for children.
Thankfully, the vast majority of kids with COVID-19 don’t get sick or show only mild flu-like symptoms. But a small percentage develop a delayed, but extremely troubling, syndrome called multisystem inflammatory syndrome in children (MIS-C). This can cause severe inflammation of the heart, lungs, kidneys, brain, and other parts of the body, coming on weeks after recovering from COVID-19. Fortunately, most kids respond to treatment and make rapid recoveries.
COVID-19’s sometimes different effects on kids likely stem not from the severity of the infection itself, but from differences in the immune response or its aftermath. Additional support for this notion comes from a new study, published in the journal Nature Medicine, that compared immune responses among children and adults with COVID-19 [1]. The study shows that the antibody responses in kids and adults with mild COVID-19 are quite similar. However, the complications seen in kids with MIS-C and adults with severe COVID-19 appear to be driven by two distinctly different types of antibodies involved in different aspects of the immune response.
The new findings come from pediatric pulmonologist Lael Yonker, Massachusetts General Hospital (MGH) Cystic Fibrosis Center, Boston, and immunologist Galit Alter, the Ragon Institute of MGH, Massachusetts Institute of Technology, and Harvard, Cambridge. Yonker runs a biorepository that collects samples from kids with cystic fibrosis. When the pandemic began, she started collecting plasma samples from children with mild COVID-19. Then, when Yonker and others began to see children hospitalized with MIS-C, she collected some plasma samples from them, too.
Using these plasma samples as windows into a child’s immune response, the research teams of Yonker and Alter detailed antibodies generated in 17 kids with MIS-C and 25 kids with mild COVID-19. They also profiled antibody responses of 60 adults with COVID-19, including 26 with severe disease.
Comparing antibody profiles among the four different groups, the researchers had expected children’s antibody responses to look quite different from those in adults. But they were in for a surprise. Adults and kids with mild COVID-19 showed no notable differences in their antibody profiles. The differences only came into focus when they compared antibodies in kids with MIS-C to adults with severe COVID-19.
In kids who develop MIS-C after COVID-19, they saw high levels of long-lasting immunoglobulin G (IgG) antibodies, which normally help to control an acute infection. Those high levels of IgG antibodies weren’t seen in adults or in kids with mild COVID-19. The findings suggest that in kids with MIS-C, those antibodies may activate scavenging immune cells, called macrophages, to drive inflammation and more severe illness.
In adults with severe COVID-19, the pattern differed. Instead of high levels of IgG antibodies, adults showed increased levels of another type of antibody, called immunoglobulin A (IgA). These IgA antibodies apparently were interacting with immune cells called neutrophils, which in turn led to the release of cytokines. That’s notable because the release of too many cytokines can cause what’s known as a “cytokine storm,” a severe symptom of COVID-19 that’s associated with respiratory distress syndrome, multiple organ failure, and other life-threatening complications.
To understand how a single virus can cause such different outcomes, studies like this one help to tease out their underlying immune mechanisms. While more study is needed to understand the immune response over time in both kids and adults, the hope is that these findings and others will help put us on the right path to discover better ways to help protect people of all ages from the most severe complications of COVID-19.
Reference:
[1] Humoral signatures of protective and pathological SARS-CoV-2 infection in children. Bartsch YC, Wang C, Zohar T, Fischinger S, Atyeo C, Burke JS, Kang J, Edlow AG, Fasano A, Baden LR, Nilles EJ, Woolley AE, Karlson EW, Hopke AR, Irimia D, Fischer ES, Ryan ET, Charles RC, Julg BD, Lauffenburger DA, Yonker LM, Alter G. Nat Med. 2021 Feb 12.
Links:
COVID-19 Research (NIH)
“NIH effort seeks to understand MIS-C, range of SARS-CoV-2 effects on children,” NIH news release, March 2, 2021.
Lael Yonker (Massachusetts General Hospital, Boston)
Alter Lab (Ragon Institute of Massachusetts General Hospital, MIT, and Harvard, Cambridge)
NIH Support: National Institute of Allergy and Infectious Diseases; National Cancer Institute
A Close-up of COVID-19 in Lung Cells
Posted on by Dr. Francis Collins
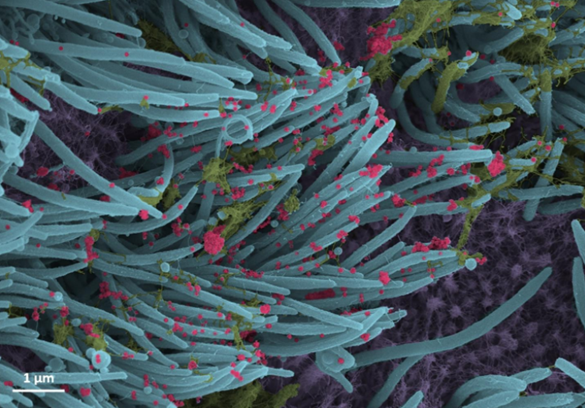
If you or a loved one have come down with SARS-CoV-2, the coronavirus responsible for COVID-19, you know it often takes hold in the respiratory system. This image offers a striking example of exactly what happens to cells in the human airway when this coronavirus infects them.
This colorized scanning electron microscope (SEM) image shows SARS-CoV-2-infected human lung cells (purple) covered in hair-like cilia (blue). Those cilia line the inner surface of the airways and help to clear mucus (yellow-green) containing dust and other debris from the lungs. Emerging from the surface of those infected airway cells are many thousands of coronavirus particles (red).
This dramatic image, published recently in the New England Journal of Medicine, comes from the lab of pediatric pulmonologist Camille Ehre, University of North Carolina at Chapel Hill. Ehre and team study mucus and how its properties change in cystic fibrosis, chronic obstructive pulmonary disease (COPD), and various other conditions that affect the lungs. These days, they’re also focusing their attention on SARS-CoV-2 and potentially new ways to block viral entry into cells of the human airway.
As part of that effort, she and her colleagues captured this snapshot of SARS-CoV-2 viruses exiting from lung cells in a lab dish. They first cultured cells from the lining of a human airway, then inoculated them with the virus. Ninety-six hours later, this is what they saw in greyscale. The vivid colors were added later by UNC medical student Cameron Morrison.
The image illustrates the astoundingly large number of viral particles that can be produced and released from infected human cells. Ehre notes that in a lab dish containing about a million human cells, they’ve witnessed the virus explode from about 1,000 particles to about 10 million in just a couple of days.
The dramatic increase in viral particles helps to explain how COVID-19 spreads so easily from the lungs to other parts of the body and—all too often—on to other individuals, especially in crowded, indoor places where people aren’t able to keep their distance. Hopefully, images like this one will help to inspire more of us this winter to avoid the crowds (especially indoors), wear masks, and wash our hands frequently.
Reference:
[1] SARS-CoV-2 infection of airway cells. Ehre C. NEJM. 2020 Sep 3;383(10):969.
Links:
Coronavirus (COVID-19) (NIH)
Camille Ehre (University of North Carolina, Chapel Hill)
Celebrating 2019 Biomedical Breakthroughs
Posted on by Dr. Francis Collins

Happy New Year! As we say goodbye to the Teens, let’s take a look back at 2019 and some of the groundbreaking scientific discoveries that closed out this remarkable decade.
Each December, the reporters and editors at the journal Science select their breakthrough of the year, and the choice for 2019 is nothing less than spectacular: An international network of radio astronomers published the first image of a black hole, the long-theorized cosmic singularity where gravity is so strong that even light cannot escape [1]. This one resides in a galaxy 53 million light-years from Earth! (A light-year equals about 6 trillion miles.)
Though the competition was certainly stiff in 2019, the biomedical sciences were well represented among Science’s “runner-up” breakthroughs. They include three breakthroughs that have received NIH support. Let’s take a look at them:
In a first, drug treats most cases of cystic fibrosis: Last October, two international research teams reported the results from phase 3 clinical trials of the triple drug therapy Trikafta to treat cystic fibrosis (CF). Their data showed Trikafta effectively compensates for the effects of a mutation carried by about 90 percent of people born with CF. Upon reviewing these impressive data, the Food and Drug Administration (FDA) approved Trikafta, developed by Vertex Pharmaceuticals.
The approval of Trikafta was a wonderful day for me personally, having co-led the team that isolated the CF gene 30 years ago. A few years later, I wrote a song called “Dare to Dream” imagining that wonderful day when “the story of CF is history.” Though we’ve still got more work to do, we’re getting a lot closer to making that dream come true. Indeed, with the approval of Trikafta, most people with CF have for the first time ever a real chance at managing this genetic disease as a chronic condition over the course of their lives. That’s a tremendous accomplishment considering that few with CF lived beyond their teens as recently as the 1980s.
Such progress has been made possible by decades of work involving a vast number of researchers, many funded by NIH, as well as by more than two decades of visionary and collaborative efforts between the Cystic Fibrosis Foundation and Aurora Biosciences (now, Vertex) that built upon that fundamental knowledge of the responsible gene and its protein product. Not only did this innovative approach serve to accelerate the development of therapies for CF, it established a model that may inform efforts to develop therapies for other rare genetic diseases.
Hope for Ebola patients, at last: It was just six years ago that news of a major Ebola outbreak in West Africa sounded a global health emergency of the highest order. Ebola virus disease was then recognized as an untreatable, rapidly fatal illness for the majority of those who contracted it. Though international control efforts ultimately contained the spread of the virus in West Africa within about two years, over 28,600 cases had been confirmed leading to more than 11,000 deaths—marking the largest known Ebola outbreak in human history. Most recently, another major outbreak continues to wreak havoc in northeastern Democratic Republic of Congo (DRC), where violent civil unrest is greatly challenging public health control efforts.
As troubling as this news remains, 2019 brought a needed breakthrough for the millions of people living in areas susceptible to Ebola outbreaks. A randomized clinical trial in the DRC evaluated four different drugs for treating acutely infected individuals, including an antibody against the virus called mAb114, and a cocktail of anti-Ebola antibodies referred to as REGN-EB3. The trial’s preliminary data showed that about 70 percent of the patients who received either mAb114 or the REGN-EB3 antibody cocktail survived, compared with about half of those given either of the other two medicines.
So compelling were these preliminary results that the trial, co-sponsored by NIH’s National Institute of Allergy and Infectious Diseases (NIAID) and the DRC’s National Institute for Biomedical Research, was halted last August. The results were also promptly made public to help save lives and stem the latest outbreak. All Ebola patients in the DRC treatment centers now are treated with one or the other of these two options. The trial results were recently published.
The NIH-developed mAb114 antibody and the REGN-EB3 cocktail are the first therapeutics to be shown in a scientifically rigorous study to be effective at treating Ebola. This work also demonstrates that ethically sound clinical research can be conducted under difficult conditions in the midst of a disease outbreak. In fact, the halted study was named Pamoja Tulinde Maisha (PALM), which means “together save lives” in Kiswahili.
To top off the life-saving progress in 2019, the FDA just approved the first vaccine for Ebola. Called Ervebo (earlier rVSV-ZEBOV), this single-dose injectable vaccine is a non-infectious version of an animal virus that has been genetically engineered to carry a segment of a gene from the Zaire species of the Ebola virus—the virus responsible for the current DRC outbreak and the West Africa outbreak. Because the vaccine does not contain the whole Zaire virus, it can’t cause Ebola. Results from a large study in Guinea conducted by the WHO indicated that the vaccine offered substantial protection against Ebola virus disease. Ervebo, produced by Merck, has already been given to over 259,000 individuals as part of the response to the DRC outbreak. The NIH has supported numerous clinical trials of the vaccine, including an ongoing study in West Africa.
Microbes combat malnourishment: Researchers discovered a few years ago that abnormal microbial communities, or microbiomes, in the intestine appear to contribute to childhood malnutrition. An NIH-supported research team followed up on this lead with a study of kids in Bangladesh, and it published last July its groundbreaking finding: that foods formulated to repair the “gut microbiome” helped malnourished kids rebuild their health. The researchers were able to identify a network of 15 bacterial species that consistently interact in the gut microbiomes of Bangladeshi children. In this month-long study, this bacterial network helped the researchers characterize a child’s microbiome and/or its relative state of repair.
But a month isn’t long enough to determine how the new foods would help children grow and recover. The researchers are conducting a similar study that is much longer and larger. Globally, malnutrition affects an estimated 238 million children under the age 5, stunting their normal growth, compromising their health, and limiting their mental development. The hope is that these new foods and others adapted for use around the world soon will help many more kids grow up to be healthy adults.
Measles Resurgent: The staff at Science also listed their less-encouraging 2019 Breakdowns of the Year, and unfortunately the biomedical sciences made the cut with the return of measles in the U.S. Prior to 1963, when the measles vaccine was developed, 3 to 4 million Americans were sickened by measles each year. Each year about 500 children would die from measles, and many more would suffer lifelong complications. As more people were vaccinated, the incidence of measles plummeted. By the year 2000, the disease was even declared eliminated from the U.S.
But, as more parents have chosen not to vaccinate their children, driven by the now debunked claim that vaccines are connected to autism, measles has made a very preventable comeback. Last October, the Centers for Disease Control and Prevention (CDC) reported an estimated 1,250 measles cases in the United States at that point in 2019, surpassing the total number of cases reported annually in each of the past 25 years.
The good news is those numbers can be reduced if more people get the vaccine, which has been shown repeatedly in many large and rigorous studies to be safe and effective. The CDC recommends that children should receive their first dose by 12 to 15 months of age and a second dose between the ages of 4 and 6. Older people who’ve been vaccinated or have had the measles previously should consider being re-vaccinated, especially if they live in places with low vaccination rates or will be traveling to countries where measles are endemic.
Despite this public health breakdown, 2019 closed out a memorable decade of scientific discovery. The Twenties will build on discoveries made during the Teens and bring us even closer to an era of precision medicine to improve the lives of millions of Americans. So, onward to 2020—and happy New Year!
Reference:
[1] 2019 Breakthrough of the Year. Science, December 19, 2019.
NIH Support: These breakthroughs represent the culmination of years of research involving many investigators and the support of multiple NIH institutes.
How Mucus Tames Microbes
Posted on by Dr. Francis Collins

Most of us think of mucus as little more than slimy and somewhat yucky stuff that’s easily ignored until you come down with a cold like the one I just had. But, when it comes to our health, there’s much more to mucus than you might think.
Mucus covers the moist surfaces of the human body, including the eyes, nostrils, lungs, and gastrointestinal tract. In fact, the average person makes more than a liter of mucus each day! It houses trillions of microbes and serves as a first line of defense against the subset of those microorganisms that cause infections. For these reasons, NIH-funded researchers, led by Katharina Ribbeck, Massachusetts Institute of Technology, Cambridge, are out to gain a greater understanding of the biology of healthy mucus—and then possibly use that knowledge to develop new therapeutics.
Ribbeck’s team used a scanning electron microscope to take the image of mucus you see above. You’ll notice right away that mucus doesn’t look like simple slime at all. In fact, if you could zoom into this complex web, you’d discover it’s made up of mucin proteins and glycans, which are sugar molecules that resemble bottle brushes.
Ribbeck and her colleagues recently discovered that the glycans in healthy mucus play a long-overlooked role in “taming” bacteria that might make us ill [1]. This work builds on their previous findings that mucus interferes with bacterial behavior, preventing these bugs from attaching to surfaces and communicating with each other [2].
In their new study, published in Nature Microbiology, Ribbeck, lead author Kelsey Wheeler, and their colleagues studied mucus and its interactions with Pseudomonas aeruginosa. This bacterium is a common cause of serious lung infections in people with cystic fibrosis or compromised immune systems.
The researchers found that in the presence of glycans, P. aeruginosa was rendered less harmful and infectious. The bacteria also produced fewer toxins. The findings show that it isn’t just that microbes get trapped in a tangled web within mucus, but rather that glycans have a special ability to moderate the bugs’ behavior. The researchers also have evidence of similar interactions between mucus and other microorganisms, such as those responsible for yeast infections.
The new study highlights an intriguing strategy to tame, rather than kill, bacteria to manage infections. In fact, Ribbeck views mucus and its glycans as a therapeutic gold mine. She hopes to apply what she’s learned to develop artificial mucus as an anti-microbial therapeutic for use inside and outside the body. Not bad for a substance that you might have thought was nothing more than slimy stuff.
References:
[1] Mucin glycans attenuate the virulence of Pseudomonas aeruginosa in infection. Wheeler KM, Cárcamo-Oyarce G, Turner BS, Dellos-Nolan S, Co JY, Lehoux S, Cummings RD, Wozniak DJ, Ribbeck K. Nat Microbiol. 2019 Oct 14.
[2] Mucins trigger dispersal of Pseudomonas aeruginosa biofilms. Co JY, Cárcamo-Oyarce, Billings N, Wheeler KM, Grindy SC, Holten-Andersen N, Ribbeck K. NPJ Biofilms Microbiomes. 2018 Oct 10;4:23.
Links:
Cystic Fibrosis (National Heart, Lung, and Blood Institute/NIH)
Video: Chemistry in Action—Katharina Ribbeck (YouTube)
Katharina Ribbeck (Massachusetts Institute of Technology, Cambridge)
NIH Support: National Institute of Biomedical Imaging and Bioengineering; National Institute of Environmental Health Sciences; National Institute of General Medical Sciences; National Institute of Allergy and Infectious Diseases
Singing “Dare to Dream”
Posted on by Dr. Francis Collins
Dare to Dream: The Long Road to Targeted Therapies for Cystic Fibrosis
Posted on by Dr. Francis Collins

When your world has been touched by a life-threatening disease, it’s hard to spend a lot of time dreaming about the future. But that’s exactly what Jenny, an 8-year-old girl with cystic fibrosis (CF), did 30 years ago upon hearing the news that I and my colleagues in Ann Arbor and Toronto had discovered the gene for CF [1,2]. Her upbeat diary entry, which you can read above, is among the many ways in which people with CF have encouraged researchers on the long and difficult road toward achieving our shared dream of effective, molecularly targeted therapies for one of the nation’s most common potentially fatal recessive genetic diseases, affecting more than 30,000 individuals in the United States [3].
Today, I’m overjoyed to say that this dream finally appears to have come true for about 90 percent of people with CF. In papers in the New England Journal of Medicine and The Lancet [4,5], two international teams, including researchers partly supported by NIH, report impressive results from phase 3 clinical trials of a triple drug therapy for individuals with CF and at least one copy of Phe508del, the most common CF-causing mutation. And Jenny happens to be among those who now stand to benefit from this major advance.
Now happily married and living in Colorado, Jenny is leading an active life, writing a children’s book and trying to keep up with her daughter Pippa Lou, whom you see with her in the photo above. In a recent email to me, her optimistic outlook continues to shine through: “I have ALWAYS known in my heart that CF will be cured during my lifetime and I have made it my goal to be strong and ready for that day when it comes. None of the advancements in care would be what they are without you.”
But there are a great many more people who need to be recognized and thanked. Such advances were made possible by decades of work involving a vast number of other researchers, many funded by NIH, as well as by more than two decades of visionary and collaborative efforts between the Cystic Fibrosis Foundation and Aurora Biosciences (now, Vertex Pharmaceuticals) that built upon that fundamental knowledge of the responsible gene and its protein product. Not only did this innovative approach serve to accelerate the development of therapies for CF, it established a model that may inform efforts to develop therapies for other rare genetic diseases.
To understand how the new triple therapy works, one first needs to understand some things about the protein affected by CF, the cystic fibrosis transmembrane regulator (CFTR). In healthy people, CFTR serves as a gated channel for chloride ions in the cell membrane, regulating the balance of salt and water in the lungs, pancreas, sweat glands, and other organ systems.
People with the most common CF-causing Phe508del mutation produce a CFTR protein with two serious problems: misfolding that often results in the protein becoming trapped in the cell’s factory production line called the endoplasmic reticulum; and deficient activation of any protein that does manage to reach its proper location in the cell membrane. Consequently, an effective therapy for such people needs to include drugs that can correct the CFTR misfolding, along with those than can activate, or potentiate, the function of CFTR when it reaches the cell membrane.
The new triple combination therapy, which was developed by Vertex Pharmaceuticals and recently approved by the Food and Drug Administration (FDA) [6], is elexacaftor-tezacaftor-ivacaftor (two correctors and one potentiator). This approach builds upon the success of ivacaftor monotherapy, approved by the FDA in 2012 for rare CF-causing mutations; and tezacaftor-ivacaftor dual therapy, approved by the FDA in 2018 for people with two copies of the Phe508del mutation.
Specifically, the final results from two Phase 3 multi-center, randomized clinical trials demonstrated the safety and efficacy of the triple combination therapy for people with either one or two copies of the Phe508del mutation—which represents about 90 percent of people with CF. Patients in both trials experienced striking improvements in a key measure of lung capacity (forced expiratory volume in 1 second) and in sweat chloride levels, which show if the drugs are working throughout the body. In addition, the triple therapy was generally safe and well tolerated, with less than 1 percent of patients discontinuing the treatment due to adverse effects.
This is wonderful news! But let’s be clear—we are not yet at our journey’s end when it comes to realizing the full dream of defeating CF. More work remains to be done to help the approximately 10 percent of CF patients whose mutations result in the production of virtually no CFTR protein, which means there is nothing for current drugs to correct or activate.
Beyond that, wouldn’t it be great if biomedical science could figure out a way to permanently cure CF, perhaps using nonheritable gene editing, so no one needs to take drugs at all? It’s a bold dream, but look how far a little dreaming, plus a lot of hard work, has taken us so far in Jenny’s life.
In closing, I’d like to leave you with the chorus of a song, called “Dare to Dream,” that I wrote shortly after we identified the CF gene. I hope the words inspire not only folks affected by CF, but everyone who is looking to NIH-supported research for healing and hope.
Dare to dream, dare to dream,
All our brothers and sisters breathing free.
Unafraid, our hearts unswayed,
‘Til the story of CF is history.
References:
[1]. Identification of the cystic fibrosis gene: chromosome walking and jumping. Rommens JM, Iannuzzi MC, Kerem B, et al. Science 1989; 245:1059-1065.
[2]. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Riordan JR, Rommens JM, Kerem B, et al. Science 1989; 245:1066-73. Erratum in: Science 1989; 245:1437.
[3] Realizing the Dream of Molecularly Targeted Therapies for Cystic Fibrosis. Collins, FS. N Engl J Med. 2019 Oct 31. [Epub ahead of print]
[4]. Elexacaftor-Tezacaftor-Ivacaftor for CF with a Single Phe508del Mutation. Middleton P, Mall M, Drevinek P, et al.N Engl J Med. 2019 Oct 31. [Epub ahead of print]
[5] Efficacy and safety of the elexacaftor/tezacaftor/ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Heijerman H, McKone E, Downey D, et al. Lancet. 2019 Oct 31. [Epub ahead of print]
[6] FDA approves new breakthrough therapy for cystic fibrosis. FDA News Release, Oct. 21, 2019.
Links:
Cystic Fibrosis (Genetics Home Reference/National Library of Medicine/NIH)
Research Milestones (Cystic Fibrosis Foundation, Bethesda, MD)
Wheezie Stevens in “Bubbles Can’t Hold Rain,” by Jennifer K. McGlincy
NIH Support: National, Heart, Lung and Blood Institute; National Institute of Diabetes and Digestive and Kidney Diseases; National Center for Advancing Translational Sciences
Thirtieth Anniversary of Cystic Fibrosis Gene Discovery
Posted on by Dr. Francis Collins
2018 Warren Alpert Foundation Prize Symposium
Posted on by Dr. Francis Collins
I was deeply honored to be among the five recipients of the 2018 Warren Alpert Foundation Prize. All five recipients were recognized for their discoveries in contributing to the development of life-altering treatments for cystic fibrosis (CF). The other recipients were: Paul Negulescu, Vertex Pharmaceuticals, Boston; Bonnie Ramsey, University of Washington School of Medicine and Seattle Children’s Research Institute; Lap-Chee Tsui, The Academy of Sciences of Hong Kong; Michael Welsh, University of Iowa, Iowa City. We were recognized at an afternoon symposium titled Cystic Fibrosis: From Gene Discovery to Basic Biology to Precision Medicines. The symposium was held at Harvard Medical School, Boston, on October 4, 2018. The video posted here shows my presentation that afternoon. But if you would like to see more, there is a full video of this fantastic symposium.
Another Milestone in the Cystic Fibrosis Journey
Posted on by Dr. Francis Collins

Caption: Two-year-old Avalyn is among the cystic fibrosis patients who may be helped by targeted drugs.
Credit: Brittany Mahoney
As NIH Director, I often hear stories of how people with serious diseases—from arthritis to Zika infection—are benefitting from the transformational power of NIH’s investments in basic science. Today, I’d like to share one such advance that I find particularly exciting: news that a combination of three molecularly targeted drugs may finally make it possible to treat the vast majority of patients with cystic fibrosis (CF), our nation’s most common genetic disease.
First, a bit of history! The first genetic mutation that causes CF was discovered by a collaborative effort between my own research lab at the University of Michigan, Ann Arbor, and colleagues at the Hospital for Sick Children in Toronto—more than 25 years ago [1]. Years of hard work, supported by the National Institutes of Health and the Cystic Fibrosis Foundation, painstakingly worked out the normal function of the protein that is altered in CF, called the cystic fibrosis transmembrane regulator (CFTR). Very recently new technologies, such as cryo-EM, have given researchers the ability to map the exact structure of the protein involved in CF.
Among the tens of thousands of CF patients who stand to benefit from the next generation of targeted drugs is little Avalyn Mahoney of Cardiff by the Sea, CA. Just a few decades ago, a kid like Avalyn—who just turned 2 last month—probably wouldn’t have made it beyond her teens. But today the outlook is far brighter for her and so many others, thanks to recent advances that build upon NIH-supported basic research.
Next Page