clinical trials
RECOVER: What Clinical Research Comes Next for Helping People with Long COVID

“I connected with RECOVER to be a part of the answers that I was looking for when I was at my worst.” Long COVID patient and RECOVER representative, Nitza Rochez (Bronx, NY)
People, like Nitza Rochez, who are living with Long COVID—the wide-ranging health issues that can follow an infection with SARS-CoV-2, the coronavirus that causes COVID-19—experience disabling symptoms with significant physical, emotional and financial consequences.
The NIH has been engaging and listening to Nitza and others living with Long COVID even before the start of its Researching COVID to Enhance Recovery (RECOVER) Initiative. But now, with the launch of RECOVER, patients and those with affected family or community members have joined researchers, clinicians, and experts in their efforts to unlock the mysteries of Long COVID. All have come together to understand what causes the condition, identify who is most at risk, and determine how to prevent and treat it.
RECOVER is unprecedented in its size and scope as the most-diverse, deeply characterized cohort of Long COVID patients. We’ve enlisted the help of many patient volunteers, who have enrolled in observational studies designed to help researchers learn as much as possible about people who have Long COVID.
Indeed, thousands of research participants are now providing health information and undergoing in-depth medical evaluations and tests, enabling investigators to look for trends. Additionally, studies of millions of electronic medical records are providing insights about those who have received care during the pandemic. More than 40 studies are being conducted to identify the causes of disease, potential biomarkers of Long COVID, and new therapeutic targets.
In all, RECOVER’s research assets are voluminous. They involve invaluable contributions from many people and communities, including research volunteers, research investigators, and clinical specialists. In addition, millions of health records and numerous related tissues and specimens are being analyzed for possible leads.
At the center of it all is the National Community Engagement Group (NCEG). The NCEG is comprised of people living with Long COVID and those representing others living with the condition, and it is truly instrumental to the initiative’s progress in understanding how and why SARS-CoV-2 impacts people in different ways. It’s also helping researchers learn why some people recover while others do not.
So far, we’ve learned that people hospitalized with COVID-19 are twice as likely to have Long COVID than those who were not hospitalized for infection. We’ve also learned that members of racial and ethnic minority groups with Long COVID were more likely to have been hospitalized with COVID-19.
Similarly, disparities in Long COVID exist within those living in areas with particular environmental exposures [1], and those who were already burdened by other diseases and conditions—such as diabetes and chronic pulmonary disease [2]. We’ve also discovered that the certain types of symptoms of Long COVID are consistent among patients regardless of which SARS-CoV-2 variant caused their initial infection. Yet, people infected with the earlier variants have a higher number of symptoms than those infected with more recent variants.
Patient experiences have guided and will continue to guide the study designs and trajectory of RECOVER. Now, fueled by the knowledge that we have gained, RECOVER is preparing to advance to the next phase of discovery—testing interventions in clinical trials to see if they can help people with Long COVID.
To prepare, we are beginning to identify potential clinical trial sites. This important step will help us to find the right places with the right staff and capabilities for enrolling the appropriate patient populations needed to implement the studies. We’ll ensure that the public knows when these upcoming clinical trials are ready to enroll.
Of course, the design of these RECOVER clinical trials will be critical, and insights gained from patients have been key in this process. Results from RECOVER study questionnaires, surveys, and discussions with people experiencing Long COVID identified symptom clusters considered to be the most significant and burdensome to patients. These include sleep disorders, “brain fog” (trouble thinking clearly), exercise intolerance and fatigue, and nervous system dysfunction affecting people’s ability to regulate normal body functions like heart rate and body temperature.
These patient observations have effectively guided the design of the clinical trials that will evaluate whether certain interventions and therapies can help alleviate symptoms that are part of these specific clusters. We’re excited to be advancing toward this phase of the initiative and, again, are very grateful to patient representatives like Nitza, quoted above, for getting us to this phase.
Effective evaluation of those treatments will be important, too. Early in the pandemic, while many clinical trials were launching, most were not large enough or did not have the appropriate objectives to define effective treatments for acute COVID-19. This left clinicians with few clear options when faced with patients needing help.
Learning from this experience, the RECOVER trials will be harmonized to ensure coordinated and efficient evaluation of interventions—in other words, all potential therapies will be using the same protocols platforms and the same data elements. This consistency accelerates our understanding and strengthens the certainty of findings.
Given the widespread and diverse impact that the virus has on the body, it is highly likely that more than one treatment will be needed for each kind of patient experience. Finding solutions for everyone—people of all races, ethnicities, genders, ages, and geographic locations—is paramount.
RECOVER patient representative, Juan Lewis, of San Antonio shared with us, “In April 2020, I was fighting for my life, and today I fight for my quality of life. COVID impacted me physically, mentally, socially, and financially.”
For people like Juan who are experiencing debilitating Long COVID symptoms, we know that finding answers as quickly as possible is critical. As we look ahead to the next 12 months, we’ll continue the studies evaluating the underlying causes, risk factors, and outcomes of Long Covid, and we anticipate significant scientific progress on research leading to Long COVID treatments.
Keep an eye on the RECOVER website for updates on our progress, and published findings.
References:
[1] Identifying environmental risk factors for post-acute sequelae of SARS-CoV-2 infection: An EHR-based cohort study from the recover program. Zhang Y, Hu H, Fokaidis V, V CL, Xu J, Zang C, Xu Z, Wang F, Koropsak M, Bian J, Hall J, Rothman RL, Shenkman EA, Wei WQ, Weiner MG, Carton TW, Kaushal R. Environ Adv. 2023 Apr;11:100352.
[2] Identifying who has long COVID in the USA: a machine learning approach using N3C data. Pfaff ER, Girvin AT, Bennett TD, Bhatia A, Brooks IM, Deer RR, Dekermanjian JP, Jolley SE, Kahn MG, Kostka K, McMurry JA, Moffitt R, Walden A, Chute CG, Haendel MA; N3C Consortium. Lancet Digit Health. 2022 Jul;4(7):e532-e541.
Links:
RECOVER: Researching COVID to Enhance Recovery
Long COVID: Ask NIH Leader about Latest Research (YouTube)
NIH Builds Large Nationwide Study Population of Tens of Thousands to Support Research on Long-Term Effects of COVID-19, NIH News Release, September 15, 2021
Understanding Long-Term COVID-19 Symptoms and Enhancing Recovery, NIH Director’s Blog, October 4, 2022.
NIH RECOVER Research Identifies Potential Long COVID Disparities. NIH News Release, February 16, 2023.
NIH RECOVER Listening Session, June 2021 (NIH Videocast)
NIH RECOVER Listening Session: Understanding Long COVID Across Communities of Color and Those Hardest Hit by COVID, January 21, 2022 (NIH Videocast)
Note: Dr. Lawrence Tabak, who performs the duties of the NIH Director, has asked the heads of NIH’s Institutes, Centers, and Offices to contribute occasional guest posts to the blog to highlight some of the interesting science that they support and conduct. This is the 25th in the series of NIH guest posts that will run until a new permanent NIH director is in place.
Gratitude for Biomedical Progress and All Those Who Make It Possible
Posted on by Lawrence Tabak, D.D.S., Ph.D.

It’s good for our health to eat right, exercise, and get plenty of rest. Still, many other things contribute to our sense of wellbeing, including making it a point to practice gratitude whenever we can. With this in mind, I can’t think of a better time than Thanksgiving to recognize just a few of the many reasons that I—and everyone who believes in the mission of the National Institutes of Health (NIH)—have to be grateful.
First, I’m thankful for the many enormously talented people with whom I’ve worked over the past year while performing the duties of the NIH Director. Particular thanks go to those on my immediate team within the Office of the Director. I could not have taken on this challenge without their dedicated support.
I’m also gratified by the continued enthusiasm and support for biomedical research from so many different corners of our society. This includes the many thousands of unsung, patient partners who put their time, effort, and, in some cases, even their lives on the line for the sake of medical progress and promising treatment advances. Without them, clinical research—including the most pivotal clinical trials—simply wouldn’t be possible.
I am most appreciative of the continuing efforts at NIH and across the broader biomedical community to further enable diversity, equity, inclusion, and accessibility within the biomedical research workforce and in all the work that NIH supports.
High on my Thanksgiving list is the widespread availability of COVID-19 bivalent booster shots. These boosters not only guard against older strains of the coronavirus, but also broaden immunity to the newer Omicron variant and its many subvariants. I’m also tremendously grateful for everyone who has—or soon will—get boosted to protect yourself, your loved ones, and your communities as the winter months fast approach.
Another big “thank you” goes out to all the researchers studying Long COVID, the complex and potentially debilitating constellation of symptoms that strikes some people after recovery from COVID-19. I look forward to more answers as this work continues and we certainly couldn’t do it without our patient partners.
I’d also like to express my appreciation for the NIH’s institute and center directors who’ve contributed to the NIH Director’s Blog to showcase NIH’s broad and diverse portfolio of promising research.
Finally, a special thanks to all of you who read this blog. As you gather with family and friends to celebrate this Thanksgiving holiday, I hope the time you spend here gives you a few more reasons to feel grateful and appreciate the importance of NIH in turning scientific discovery into better health for all.
Understanding Long-Term COVID-19 Symptoms and Enhancing Recovery
Posted on by Walter J. Koroshetz, M.D., National Institute of Neurological Disorders and Stroke

We are in the third year of the COVID-19 pandemic, and across the world, most restrictions have lifted, and society is trying to get back to “normal.” But for many people—potentially millions globally—there is no getting back to normal just yet.
They are still living with the long-term effects of a COVID-19 infection, known as the post-acute sequelae of SARS-CoV-2 infection (PASC), including Long COVID. These people continue to experience debilitating fatigue, shortness of breath, pain, difficulty sleeping, racing heart rate, exercise intolerance, gastrointestinal and other symptoms, as well as cognitive problems that make it difficult to perform at work or school.
This is a public health issue that is in desperate need of answers. Research is essential to address the many puzzling aspects of Long COVID and guide us to effective responses that protect the nation’s long-term health.
For the past two years, NIH’s National Heart, Lung, and Blood Institute (NHLBI), the National Institute of Allergy and Infectious Diseases (NIAID), and my National Institute of Neurological Disorders and Stroke (NINDS) along with several other NIH institutes and the office of the NIH Director, have been leading NIH’s Researching COVID to Enhance Recovery (RECOVER) initiative, a national research program to understand PASC.
The initiative studies core questions such as why COVID-19 infections can have lingering effects, why new symptoms may develop, and what is the impact of SARS-CoV-2, the virus that causes COVID-19, on other diseases and conditions? Answering these fundamental questions will help to determine the underlying biologic basis of Long COVID. The answers will also help to tell us who is at risk for Long COVID and identify therapies to prevent or treat the condition.
The RECOVER initiative’s wide scope of research is also unprecedented. It is needed because Long COVID is so complex, and history indicates that similar post infectious conditions have defied definitive explanation or effective treatment. Indeed, those experiencing Long COVID report varying symptoms, making it highly unlikely that a single therapy will work for everyone, underscoring the need to pursue multiple therapeutic strategies.
To understand Long COVID fully, hundreds of RECOVER investigators are recruiting more than 17,000 adults (including pregnant people) and more than 18,000 children to take part in cohort studies. Hundreds of enrolling sites have been set up across the country. An autopsy research cohort will also provide further insight into how COVID-19 affects the body’s organs and tissues.
In addition, researchers will analyze electronic health records from millions of people to understand how Long COVID and its symptoms change over time. The RECOVER initiative is also utilizing consistent research protocols across all the study sites. The protocols have been carefully developed with input from patients and advocates, and they are designed to allow for consistent data collection, improve data sharing, and help to accelerate the pace of research.
From the very beginning, people suffering from Long COVID have been our partners in RECOVER. Patients and advocates have contributed important perspectives and provided valuable input into the master protocols and research plans.
Now, with RECOVER underway, individuals with Long COVID, their caregivers, and community members continue to serve a critical role in the Initiative. The National Community Engagement Group (NCEG) has been established to make certain that RECOVER meets the needs of all people affected by Long COVID. The RECOVER Patient and Community Engagement Strategy outlines all the approaches that RECOVER is using to engage with and gather input from individuals impacted by Long COVID.
The NIH recently made more than 40 awards to improve understanding of the underlying biology and pathology of Long COVID. There have already been several important findings published by RECOVER scientists.
For example, in a recent study published in the journal Lancet Digital Health, RECOVER investigators used machine learning to comb through electronic health records to look for signals that may predict whether someone has Long COVID [1]. As new findings, tools, and technologies continue to emerge that help advance our knowledge of the condition, the RECOVER Research Review (R3) Seminar Series will provide a forum for researchers and our partners with up-to-date information about Long COVID research.
It is important to note that post-viral conditions are not a new concept. Many, but not all, of the symptoms reported in Long COVID, including fatigue, post-exertional malaise, chronic musculoskeletal pain, sleep disorders, postural orthostatic tachycardia (POTS), and cognitive issues, overlap with myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS).
ME/CFS is a serious disease that can occur following infection and make people profoundly sick for decades. Like Long COVID, ME/CFS is a heterogenous condition that does not affect everybody in the same way, and the knowledge gained through research on Long COVID may also positively impact the understanding, treatment, and prevention of POTS, ME/CFS, and other chronic diseases.
Unlike other post-viral conditions, people who experience Long COVID were all infected by the same virus—albeit different variants—at a similar point in time. This creates a unique opportunity for RECOVER researchers to study post-viral conditions in real-time.
The opportunity enables scientists to study many people simultaneously while they are still infected to monitor their progress and recovery, and to try to understand why some individuals develop ongoing symptoms. A better understanding of the transition from acute to chronic disease may offer an opportunity to intervene, identify who is at risk of the transition, and develop therapies for people who experience symptoms long after the acute infection has resolved.
The RECOVER initiative will soon announce clinical trials, leveraging data from clinicians and patients in which symptom clusters were identified and can be targeted by various interventions. These trials will investigate therapies that are indicated for other non-COVID conditions and novel treatments for Long COVID.
Through extensive collaboration across the multiple NIH institutes and offices that contribute to the RECOVER effort, our hope is critical answers will emerge soon. These answers will help us to recognize the full range of outcomes and needs resulting from PASC and, most important, enable many people to make a full recovery from COVID-19. We are indebted to the over 10,000 subjects who have already enrolled in RECOVER. Their contributions and the hard work of the RECOVER investigators offer hope for the future to the millions still suffering from the pandemic.
Reference:
[1] Identifying who has long COVID in the USA: a machine learning approach using N3C data. Pfaff ER, Girvin AT, Bennett TD, Bhatia A, Brooks IM, Deer RR, Dekermanjian JP, Jolley SE, Kahn MG, Kostka K, McMurry JA, Moffitt R, Walden A, Chute CG, Haendel MA; N3C Consortium. Lancet Digit Health. 2022 Jul;4(7):e532-e541.
Links:
COVID-19 Research (NIH)
Long COVID (NIH)
RECOVER: Researching COVID to Enhance Recovery (NIH)
“NIH builds large nationwide study population of tens of thousands to support research on long-term effects of COVID-19,” NIH News Release, September 15, 2021.
Director’s Messages (National Institute of Neurological Disorders and Stroke/NIH)
Note: Dr. Lawrence Tabak, who performs the duties of the NIH Director, has asked the heads of NIH’s Institutes and Centers (ICs) to contribute occasional guest posts to the blog to highlight some of the interesting science that they support and conduct. This is the 18th in the series of NIH IC guest posts that will run until a new permanent NIH director is in place.
Tuberculosis: An Ancient Disease in Need of Modern Scientific Tools
Posted on by Anthony S. Fauci, M.D., National Institute of Allergy and Infectious Diseases

Although COVID-19 has dominated our attention for the past two years, tuberculosis (TB), an ancient scourge, remains a dominating infectious disease globally, with an estimated 10 million new cases and more than 1.3 million deaths in 2020. TB disproportionately afflicts the poor and has long been the leading cause of death in people living with HIV.
Unfortunately, during the global COVID-19 pandemic, recent gains in TB control have been stalled or reversed. We’ve seen a massive drop in new TB diagnoses, reflecting poor access to care and an uptick in deaths in 2020 [1].
We are fighting TB with an armory of old weapons inferior to those we have for COVID-19. The Bacillus Calmette–Guérin (BCG) vaccine, the world’s only licensed TB vaccine, has been in use for more than 100 years. While BCG is somewhat effective at preventing TB meningitis in children, it provides more limited durable protection against pulmonary TB in children and adults. More effective vaccination strategies to prevent infection and disease, decrease relapse rates, and shorten durations of treatment are desperately needed to reduce the terrible global burden of TB.
In this regard, over the past five years, several exciting research advances have generated new optimism in the field of TB vaccinology. Non-human primate studies conducted at my National Institute of Allergy and Infectious Diseases’ (NIAID) Vaccine Research Center and other NIAID-funded laboratories have demonstrated that effective immunity against infection is achievable and that administering BCG intravenously, rather than under the skin as it currently is given, is highly protective [2].
Results from a phase 2 trial testing BCG revaccination in adolescents at high risk of TB infection suggested this approach could help prevent TB [3]. In addition, a phase 2 trial of an experimental TB vaccine based on the recombinant protein M72 and an immune-priming adjuvant, AS01, also showed promise in preventing active TB disease in latently infected adults [4].
Both candidates are now moving on to phase 3 efficacy trials. The encouraging results of these trials, combined with nine other candidates currently in phase 2 or 3 studies [5], offer new hope that improved vaccines may be on the horizon. The NIAID is working with a team of other funders and investigators to analyze the correlates of protection from these studies to inform future TB vaccine development.
Even with these exciting developments, it is critical to accelerate our efforts to enhance and diversify the TB vaccine pipeline by addressing persistent basic and translational research gaps. To this end, NIAID has several new programs. The Immune Protection Against Mtb Centers are taking a multidisciplinary approach to integrate animal and human data to gain a comprehensive understanding of the immune responses required to prevent TB infection and disease.
This spring, NIAID will fund awards under the Innovation for TB Vaccine Discovery program that will focus on the discovery and early evaluation of novel TB vaccine candidates with the goal of diversifying the TB vaccine pipeline. Later this year, the Advancing Vaccine Adjuvant Research for TB program will systematically assess combinations of TB immunogens and adjuvants. Finally, NIAID’s well-established clinical trials networks are planning two new clinical trials of TB vaccine candidates.
As we look to the future, we must apply the lessons learned in the development of the COVID-19 vaccines to longstanding public health challenges such as TB. COVID-19 vaccine development was hugely successful due to the use of novel vaccine platforms, structure-based vaccine design, community engagement for rapid clinical trial enrollment, real-time data sharing with key stakeholders, and innovative trial designs.
However, critical gaps remain in our armamentarium. These include the harnessing the immunology of the tissues that line the respiratory tract to design vaccines more adept at blocking initial infection and transmission, employing thermostable formulations and novel delivery systems for resource-limited settings, and crafting effective messaging around vaccines for different populations.
As we work to develop better ways to prevent, diagnose, and treat TB, we will do well to remember the great public health icon, Paul Farmer, who tragically passed away earlier this year at a much too young age. Paul witnessed firsthand the devastating consequences of TB and its drug resistant forms in Haiti, Peru, and other parts of the world.
In addition to leading efforts to improve how TB is treated, Paul provided direct patient care in underserved communities and demanded that the world do more to meet their needs. As we honor Paul’s legacy, let us accelerate our efforts to find better tools to fight TB and other diseases of global health importance that exact a disproportionate toll among the poor and underserved.
References:
[1] Global tuberculosis report 2021. WHO. October 14, 2021.
[2] Prevention of tuberculosis in macaques after intravenous BCG immunization. Darrah PA, Zeppa JJ, Maiello P, Hackney JA, Wadsworth MH,. Hughes TK, Pokkali S, Swanson PA, Grant NL, Rodgers MA, Kamath M, Causgrove CM, Laddy DJ, Bonavia A, Casimiro D, Lin PL, Klein E, White AG, Scanga CA, Shalek AK, Roederer M, Flynn JL, and Seder RA. Nature. 2020 Jan 1; 577: 95–102.
[3] Prevention of M. tuberculosis Infection with H4:IC31 vaccine or BCG revaccination. Nemes E, Geldenhuys H, Rozot V, Rutkowski KT, Ratangee F,Bilek N., Mabwe S, Makhethe L, Erasmus M, Toefy A, Mulenga H, Hanekom WA, et al. N Engl J Med 2018; 379:138-149.
[4] Final analysis of a trial of M72/AS01E vaccine to prevent tuberculosis. Tait DR, Hatherill M, Van Der Meeren O, Ginsberg AM, Van Brakel E, Salaun B, Scriba TJ, Akite EJ, Ayles HM, et al.
[5] Pipeline Report 2021: Tuberculosis Vaccines. TAG. October 2021.
Links:
Tuberculosis (National Institute of Allergy and Infectious Diseases/NIH)
NIAID Strategic Plan for Tuberculosis Research
Immune Mechanisms of Protection Against Mycobacterium tuberculosis Centers (IMPAc-TB) (NIAID)
Partners in Health (Boston, MA)
[Note: Acting NIH Director Lawrence Tabak has asked the heads of NIH’s Institutes and Centers (ICs) to contribute occasional guest posts to the blog to highlight some of the interesting science that they support and conduct. This is the seventh in the series of NIH IC guest posts that will run until a new permanent NIH director is in place.]
NCI Support for Basic Science Paves Way for Kidney Cancer Drug Belzutifan
Posted on by Norman "Ned" Sharpless, M.D., National Cancer Institute

There’s exciting news for people with von Hippel-Lindau (VHL) disease, a rare genetic disorder that can lead to cancerous and non-cancerous tumors in multiple organs, including the brain, spinal cord, kidney, and pancreas. In August 2021, the U.S. Food and Drug Administration (FDA) approved belzutifan (Welireg), a new drug that has been shown in a clinical trial led by National Cancer Institute (NCI) researchers to shrink some tumors associated with VHL disease [1], which is caused by inherited mutations in the VHL tumor suppressor gene.
As exciting as this news is, relatively few people have this rare disease. The greater public health implication of this advancement is for people with sporadic, or non-inherited, clear cell kidney cancer, which is by far the most common subtype of kidney cancer, with more than 70,000 cases and about 14,000 deaths per year. Most cases of sporadic clear cell kidney cancer are caused by spontaneous mutations in the VHL gene.
This advancement is also a great story of how decades of support for basic science through NCI’s scientists in the NIH Intramural Research Program and its grantees through extramural research funding has led to direct patient benefit. And it’s a reminder that we never know where basic science discoveries might lead.
Belzutifan works by disrupting the process by which the loss of VHL in a tumor turns on a series of molecular processes. These processes involve the hypoxia-inducible factor (HIF) transcription factor and one of its subunits, HIF-2α, that lead to tumor formation.
The unraveling of the complex relationship among VHL, the HIF pathway, and cancer progression began in 1984, when Bert Zbar, Laboratory of Immunobiology, NCI-Frederick; and Marston Linehan, NCI’s Urologic Oncology Branch, set out to find the gene responsible for clear cell kidney cancer. At the time, there were no effective treatments for advanced kidney cancer, and 80 percent of patients died within two years.
Zbar and Linehan started by studying patients with sporadic clear cell kidney cancer, but then turned their focus to investigations of people affected with VHL disease, which predisposes a person to developing clear cell kidney cancer. By studying the patients and the genetic patterns of tumors collected from these patients, the researchers hypothesized that they could find genes responsible for kidney cancer.
Linehan established a clinical program at NIH to study and manage VHL patients, which facilitated the genetic studies. It took nearly a decade, but, in 1993, Linehan, Zbar, and Michael Lerman, NCI-Frederick, identified the VHL gene, which is mutated in people with VHL disease. They soon discovered that tumors from patients with sporadic clear cell kidney cancer also have mutations in this gene.
Subsequently, with NCI support, William G. Kaelin Jr., Dana-Farber Cancer Institute, Boston, discovered that VHL is a tumor suppressor gene that, when inactivated, leads to the accumulation of HIF.
Another NCI grantee, Gregg L. Semenza, Johns Hopkins School of Medicine, Baltimore, identified HIF as a transcription factor. And Peter Ratcliffe, University of Oxford, United Kingdom, discovered that HIF plays a role in blood vessel development and tumor growth.
Kaelin and Ratcliffe simultaneously showed that the VHL protein tags a subunit of HIF for destruction when oxygen levels are high. These results collectively answered a very old question in cell biology: How do cells sense the intracellular level of oxygen?
Subsequent studies by Kaelin, with NCI’s Richard Klausner and Linehan, revealed the critical role of HIF in promoting the growth of clear cell kidney cancer. This work ultimately focused on one member of the HIF family, the HIF-2α subunit, as the key mediator of clear cell kidney cancer growth.
The fundamental work of Kaelin, Semenza, and Ratcliffe earned them the 2019 Nobel Prize in Physiology or Medicine. It also paved the way for drug discovery efforts that target numerous points in the pathway leading to clear cell kidney cancer, including directly targeting the transcriptional activity of HIF-2α with belzutifan.
Clinical trials of belzutifan, including several supported by NCI, demonstrated potent anti-cancer activity in VHL-associated kidney cancer, as well as other VHL-associated tumors, leading to the aforementioned recent FDA approval. This is an important development for patients with VHL disease, providing a first-in-class therapy that is effective and well-tolerated.
We believe this is only the beginning for belzutifan’s use in patients with cancer. A number of trials are now studying the effectiveness of belzutifan for sporadic clear cell kidney cancer. A phase 3 trial is ongoing, for example, to look at the effectiveness of belzutifan in treating people with advanced kidney cancer. And promising results from a phase 2 study show that belzutifan, in combination with cabozantinib, a widely used agent to treat kidney cancer, shrinks tumors in patients previously treated for metastatic clear cell kidney cancer [2].
This is a great scientific story. It shows how studies of familial cancer and basic cell biology lead to effective new therapies that can directly benefit patients. I’m proud that NCI’s support for basic science, both intramurally and extramurally, is making possible many of the discoveries leading to more effective treatments for people with cancer.
References:
[1] Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, Oudard S, Else T, Maranchie JK, Welsh SJ, Thamake S, Park EK, Perini RF, Linehan WM, Srinivasan R; MK-6482-004 Investigators. N Engl J Med. 2021 Nov 25;385(22):2036-2046.
[2] Phase 2 study of the oral hypoxia-inducible factor 2α (HIF-2α) inhibitor MK-6482 in combination with cabozantinib in patients with advanced clear cell renal cell carcinoma (ccRCC). Choueiri TK et al. J Clin Oncol. 2021 Feb 20;39(6_suppl): 272-272.
Links:
Von Hippel-Lindau Disease (Genetic and Rare Diseases Information Center/National Center for Advancing Translational Sciences/NIH)
Clear Cell Renal Cell Carcinoma (National Cancer Institute/NIH)
Belzutifan Approved to Treat Tumors Linked to Inherited Disorder VHL, Cancer Currents Blog, National Cancer Institute, September 21, 2021.
The Long Road to Understanding Kidney Cancer (Intramural Research Program/NIH)
[Note: Acting NIH Director Lawrence Tabak has asked the heads of NIH’s institutes and centers to contribute occasional guest posts to the blog as a way to highlight some of the cool science that they support and conduct. This is the first in the series of NIH institute and center guest posts that will run until a new permanent NIH director is in place.]
Partnership to Expand Effective Gene Therapies for Rare Diseases
Posted on by Dr. Francis Collins
Rare diseases aren’t so rare. Collectively, up to 30 million Americans, many of them children, are born with one of the approximately 7,000 known rare diseases. Most of these millions of people also share a common genetic feature: their diseases are caused by an alteration in a single gene.
Many of these alterations could theoretically be targeted with therapies designed to correct or replace the faulty gene. But there have been significant obstacles in realizing this dream. The science of gene therapy has been making real progress, but pursuing promising approaches all the way to clinical trials and gaining approval from the U.S. Food and Drug Administration (FDA) is still very difficult. Another challenge is economic: for the rarest of these conditions (which is most of them), the market is so small that most companies have no financial incentive to pursue them.
To overcome these obstacles and provide hope for those with rare diseases, we need a new way of doing things. One way to do things differently—and more efficiently—is the recently launched Bespoke Gene Therapy Consortium (BGTC). It is a bold partnership of NIH, the FDA, 10 pharmaceutical companies, several non-profit organizations, and the Foundation for the National Institutes of Health [1]. Its aim: optimize the gene therapy development process and help fill the significant unmet medical needs of people with rare diseases.
The BGTC, which is also part of NIH’s Accelerating Medicines Partnership® (AMP®), will enable the easier, faster, and cheaper pursuit of “bespoke” gene therapies, meaning made for a particular customer or user. The goal of the Consortium is to reduce the cost of gene therapy protocols and increase the likelihood of success, making it more attractive for companies to invest in rare diseases and bring treatments to patients who desperately need them.
Fortunately, there is already some precedent. The BGTC effort builds on a pilot project led by NIH’s National Center for Advancing Translational Sciences (NCATS) known as Platform Vector Gene Therapy (PaVe-GT). This pilot project has helped to develop adeno-associated viruses (AAVs), which are small benign viruses engineered in the lab to carry a therapeutic gene. They are commonly used in gene therapy-related clinical trials of rare diseases.
Since the launch of PaVe-GT two years ago, the project has helped to introduce greater efficiency to gene therapy trials for rare disease. It’s also offered a way to get around the standard one-disease-at-a-time approach to therapeutic development that has stymied progress in treating rare conditions.
The BGTC will now continue to advance in-depth understanding of basic AAV biology and develop better gene therapies for rare and also common diseases. The consortium aims to develop a standard set of analytic tests to improve the production and functional assessment of AAVs and therapeutic genes. Such tests will be broadly applicable and will bring the needed manufacturing efficiency required for developing gene therapies for very rare conditions.
The BGTC also will work toward bringing therapies sooner to individuals in need. To start, BGTC-funded research will support four to six clinical trials, each focused on a distinct rare disease. While the details haven’t yet been decided, these diseases are expected to be rare, single-gene diseases that lack gene therapies or commercial programs in development, despite having substantial groundwork in place to enable the rapid initiation of preclinical and clinical studies.
Through these trials, the BGTC will chart a path from studies in animal models of disease to human clinical trials that cuts years off the development process. This will include exploring methods to streamline regulatory requirements and processes for FDA approval of safe and effective gene therapies, including developing standardized approaches to preclinical testing.
This work promises to be a significant investment in helping people with rare diseases. The NIH and private partners will contribute approximately $76 million over five years to support BGTC-funded projects. This includes about $39.5 million from the participating NIH institutes and centers, pending availability of funds. The NCATS, which is NIH’s lead for BGTC, is expected to contribute approximately $8 million over five years.
Today, only two rare inherited conditions have FDA-approved gene therapies. The hope is this investment will raise that number and ultimately reduce the many significant challenges, including health care costs, faced by families that have a loved one with a rare disease. In fact, a recent study found that health care costs for people with a rare disease are three to five times greater than those for people without a rare disease [2]. These families need help, and BGTC offers an encouraging new way forward for them.
References:
[1] NIH, FDA and 15 private organizations join forces to increase effective gene therapies for rare diseases. NIH news release, October 27, 2021.
[2] The IDeaS initiative: pilot study to assess the impact of rare diseases on patients and healthcare systems. Tisdale, A., Cutillo, C.M., Nathan, R. et al. Orphanet J Rare Dis 16, 429 (2021).
Links:
FAQ About Rare Diseases (National Center for Advancing Translational Sciences/NIH)
Bespoke Gene Therapy Consortium (BGTC)
Platform Vector Gene Therapy (NCATS)
Accelerating Medicines Partnership® (AMP®) (NIH)
NIH Support: National Center for Advancing Translational Sciences; Eunice Kennedy Shriver National Institute of Child Health and Human Development; National Eye Institute; National Heart, Lung, and Blood Institute; National Human Genome Research Institute; National Institute of Arthritis and Musculoskeletal and Skin Diseases; National Institute of Dental and Craniofacial Research; National Institute of Mental Health; National Institute of Neurological Disorders and Stroke; National Institute on Deafness and Other Communication Disorders; and NIH’s BRAIN Initiative.
ACTIV Update: Making Major Strides in COVID-19 Therapeutic Development
Posted on by Dr. Francis Collins

Right now, many U.S. hospitals are stretched to the limit trying to help people battling serious cases of COVID-19. But as traumatic as this experience still is for patients and their loved ones, the chances of surviving COVID-19 have in fact significantly improved in the year since the start of the pandemic.
This improvement stems from several factors, including the FDA’s emergency use authorization (EUA) of a number of therapies found to be safe and effective for COVID-19. These include drugs that you may have heard about on the news: remdesivir (an antiviral), dexamethasone (a steroid), and monoclonal antibodies from the companies Eli Lilly and Regeneron.
Yet the quest to save more lives from COVID-19 isn’t even close to being finished, and researchers continue to work intensively to develop new and better treatments. A leader in this critical effort is NIH’s Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) initiative, a public-private partnership involving 20 biopharmaceutical companies, academic experts, and multiple federal agencies.
ACTIV was founded last April to accelerate drug research that typically requires more than a decade of clinical ups and downs to develop a safe, effective therapy. And ACTIV has indeed moved at unprecedented speed since its launch. Cutting through the usual red tape and working with an intense sense of purpose, the partnership took a mere matter of weeks to set up its first four clinical trials. Beyond the agents mentioned above that have already been granted an EUA, ACTIV is testing 15 additional potential agents, with several of these already demonstrating promising results.
Here’s how ACTIV works. The program relies on four expert “working groups” with specific charges:
Preclinical Working Group: Shares standardized preclinical evaluation resources and accelerate testing of candidate therapies and vaccines for clinical trials.
Therapeutics Clinical Working Group: Prioritizes therapeutic agents for testing within an adaptive master protocol strategy for clinical research.
Clinical Trial Capacity Working Group: Has developed and organized an inventory of clinical trial capacity that can serve as potential settings in which to implement effective COVID-19 clinical trials.
Vaccines Working Group: Accelerates the evaluation of vaccine candidates.
To give you just one example of how much these expert bodies have accomplished in record time, the Therapeutics Clinical Working Group got to work immediately evaluating some 400 candidate therapeutics using multiple publicly available information sources. These candidates included antivirals, host-targeted immune modulators, monoclonal antibodies (mAb), and symptomatic/supportive agents including anticoagulants. To follow up on even more new leads, the working group launched a COVID-19 Clinical & Preclinical Candidate Compound Portal, which remains open for submissions of therapeutic ideas and data.
All the candidate agents have been prioritized using rigorous scoring and assessment criteria. What’s more, the working group simultaneously developed master protocols appropriate for each of the drug classes selected and patient populations: outpatient, inpatient, or convalescent.
Through the coordinated efforts of all the working groups, here’s where we stand with the ACTIV trials:
ACTIV-1: A large-scale Phase 3 trial is enrolling hospitalized adults to test the safety and effectiveness of three medicines (cenicriviroc, abatacept, and infliximab). They are called immune modulators because they help to minimize the effects of an overactive immune response in some COVID-19 patients. This response, called a “cytokine storm,” can lead to acute respiratory distress syndrome, multiple organ failure, and other life-threatening complications.
ACTIV-2: A Phase 2/3 trial is enrolling adults with COVID-19 who are not hospitalized to evaluate the safety of multiple monoclonal antibodies (Lilly’s LY-CoV555, Brii Biosciences’s BRII-196 and BRII-198, and AstraZeneca’s AZD7442) used to block or neutralize the SARS-CoV-2 virus. The Lilly monoclonal antibody LY-CoV555 received an EUA for high risk non-hospitalized patients on November 9, 2020 and ACTIV-2 continued to test the agent in an open label study to further determine safety and efficacy in outpatients. Another arm of this trial has just started, testing inhaled, easy-to-administer interferon beta-1a treatment in adults with mild-to-moderate COVID-19 who are not hospitalized. An additional arm will test the drug camostat mesilate, a protease inhibitor that can block the TMPRSS2 host protein that is necessary for viral entry into human cells.
ACTIV-3: This Phase 3 trial is enrolling hospitalized adults with COVID-19. This study primarily aims to evaluate safety and to understand if monoclonal antibodies (AstraZeneca’s AZD7442, BRII-196 and BRII-198, and the VIR-7831 from GSK/Vir Biotechnology) and potentially other types of therapeutics can reduce time to recovery. It also aims to understand a treatment’s effect on extrapulmonary complications and respiratory dysfunction. Lilly’s monoclonal antibody LY-CoV555 was one of the first agents to be tested in this clinical trial and it was determined to not show the same benefits seen in outpatients. [Update: NIH-Sponsored ACTIV-3 Clinical Trial Closes Enrollment into Two Sub-Studies, March 4, 2021]
ACTIV-4: This trial aims to determine if various types of blood thinners, including apixaban, aspirin, and both unfractionated (UF) and low molecular weight (LMW) heparin, can treat adults diagnosed with COVID-19 and prevent life-threatening blood clots from forming. There are actually three Phase 3 trials included in ACTIV-4. One is enrolling people diagnosed with COVID-19 but who are not hospitalized; a second is enrolling patients who are hospitalized; and a third is enrolling people who are recovering from COVID-19. ACTIV-4 has already shown that full doses of heparin blood thinners are safe and effective for moderately ill hospitalized patients.
ACTIV-5: This is a Phase 2 trial testing newly identified agents that might have a major benefit to hospitalized patients with COVID-19, but that need further “proof of concept” testing before they move into a registrational Phase 3 trial. (In fact, another name for this trial is the “Big Effect Trial”.) It is testing medicines previously developed for other conditions that might be beneficial in treatment of COVID-19. The first two agents being tested are risankizumab (the result of a collaboration between Boehringer-Ingelheim), which is already FDA-approved to treat plaque psoriasis, and lenzilumab, which is under development by Humanigen to treat patients experiencing cytokine storm as part of cancer therapy.
In addition to trials conducted under the ACTIV partnership, NIH has prioritized and tested additional therapeutics in “ACTIV-associated trials.” These are NIH-funded, randomized, placebo-controlled clinical trials with one or more industry partners. Here’s a table with a comprehensive list.
Looking a bit further down the road, we also seek to develop orally administered drugs that would potentially block the replication ability of SARS-CoV-2, the coronavirus that causes COVID-19, in the earliest stages of infection. One goal would be to develop an antiviral medication for SARS-CoV-2 that acts similarly to oseltamivir phosphate (Tamiflu®), a drug used to shorten the course of the flu in people who’ve had symptoms for less than two days and to prevent the flu in asymptomatic people who may have been exposed to the influenza virus. Yet another major long-term effort of NIH and its partners will be to develop safe and effective antiviral medications that work against all coronaviruses, even those with variant genomes. (And, yes, such drugs might even cure the common cold!)
So, while our ACTIV partners and many other researchers around the globe continue to harness the power of science to end the devastating COVID-19 pandemic as soon as possible, we must also consider the lessons learned this past year, in order to prepare ourselves to respond more swiftly to future outbreaks of coronaviruses and other infectious disease threats. Our work is clearly a marathon, not a sprint.
Links:
Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) (NIH)
COVID-19 Research (NIH)
Combat COVID (U.S. Department of Health and Human Services, Washington, D.C.)
Pull Up a Chair with Dr. Freire: The COVID Conversations (Foundation for the National Institutes of Health, Bethesda, MD)
SARS-COV-2 Antiviral Therapeutics Summit Report, November 2020 (NIH)
Can Blood Thinners Keep Moderately Ill COVID-19 Patients Out of the ICU?
Posted on by Dr. Francis Collins
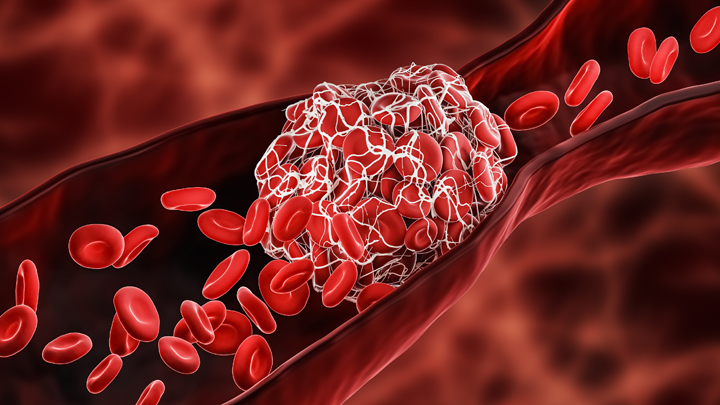
One of many troubling complications of infection with SARS-CoV-2, the coronavirus that causes COVID-19, is its ability to trigger the formation of multiple blood clots, most often in older people but sometimes in younger ones, too. It raises the question of whether and when more aggressive blood thinning treatments might improve outcomes for people hospitalized for COVID-19.
The answer to this question is desperately needed to help guide clinical practice. So, I’m happy to report interim results of three large clinical trials spanning four continents and more than 300 hospitals that are beginning to provide critical evidence on this very question [1]. While it will take time to reach a solid consensus, the findings based on more than 1,000 moderately ill patients suggest that full doses of blood thinners are safe and can help to keep folks hospitalized with COVID-19 from becoming more severely ill and requiring some form of organ support.
The results that are in so far suggest that individuals hospitalized, but not severely ill, with COVID-19 who received a full intravenous dose of the common blood thinner heparin were less likely to need vital organ support, including mechanical ventilation, compared to those who received the lower “prophylactic” subcutaneous dose. It’s important to note that these findings are in contrast to results announced last month indicating that routine use of a full dose of blood thinner for patients already critically ill and in the ICU wasn’t beneficial and may even have been harmful in some cases [2]. This is a compelling example of how critical it is to stratify patients with different severity in clinical trials—what might help one subgroup might be of no benefit, or even harmful, in another.
More study is clearly needed to sort out all the details about when more aggressive blood thinning treatment is warranted. Trial investigators are now working to make the full results available to help inform a doctor’s decisions about how to best to treat their patients hospitalized with COVID-19. It’s worth noting that these trials are overseen by independent review boards, which routinely evaluate the data and are composed of experts in ethics, biostatistics, clinical trials, and blood clotting disorders.
These clinical trials were made possible in part by the Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) public-private partnership and its ACTIV-4 Antithrombotics trials—along with similar initiatives in Canada, Australia, and the European Union. The ACTIV-4 trials are overseen by the NIH’s National Heart, Lung, and Blood institute and funded by Operation Warp Speed.
This ACTIV-4 trial is one of three Phase 3 clinical trials evaluating the safety and effectiveness of blood thinners for patients with COVID-19 [3]. Another ongoing trial is investigating whether blood thinners are beneficial for newly diagnosed COVID-19 patients who do not require hospitalization. There are also plans to explore the use of blood thinners for patients after they’ve been discharged from the hospital following a diagnosis of moderate to severe COVID-19 and to establish more precise methods for identifying which patients with COVID-19 are most at risk for developing life-threatening blood clots.
Meanwhile, research teams are exploring other potentially promising ways to repurpose existing therapeutics and improve COVID-19 outcomes. In fact, the very day that these latest findings on blood thinners were announced, another group at The Montreal Heart Institute, Canada, announced preliminary results of the international COLCORONA trial, testing the use of colchicine—an anti-inflammatory drug widely used to treat gout and other conditions—for patients diagnosed with COVID-19 [4].
Their early findings in treating patients just after a confirmed diagnosis of COVID-19 suggest that colchicine might reduce the risk of death or hospitalization compared to patients given a placebo. In the more than 4,100 individuals with a proven diagnosis of COVID-19, colchicine significantly reduced hospitalizations by 25 percent, the need for mechanical ventilation by 50 percent, and deaths by 44 percent. Still, the actual numbers of individuals represented by these percentages was small.
Time will tell whether and for which patients colchicine and blood thinners prove most useful in treating COVID-19. For those answers, we’ll have to await the analysis of more data. But the early findings on both treatment strategies come as a welcome reminder that we continue to make progress each day on such critical questions about which existing treatments can be put to work to improve outcomes for people with COVID-19. Together with our efforts to slow the spread of SARS-CoV-2, finding better ways to treat those who do get sick and prevent some of the worst outcomes will help us finally put this terrible pandemic behind us.
References:
[1] Full-dose blood thinners decreased need for life support and improved outcome in hospitalized COVID-19 patients. National Heart, Lung, and Blood Institute. January 22, 2021.
[2] NIH ACTIV trial of blood thinners pauses enrollment of critically ill COVID-19 patients. National Heart, Lung, and Blood Institute. December 22, 2020.
[3] NIH ACTIV initiative launches adaptive clinical trials of blood-clotting treatments for COVID-19. National Heart, Lung, and Blood Institute. September 10, 2020.
[4] Colchicine reduces the risk of COVID-19-related complications. The Montreal Heart Institute. January 22, 2021.
Links:
COVID-19 Research (NIH)
Combat COVID (U.S. Department of Health and Human Services, Washington, D.C.)
Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) (NIH)
NIH Support: National Heart, Lung, and Blood Institute
Celebrating the Gift of COVID-19 Vaccines
Posted on by Dr. Francis Collins

The winter holidays are traditionally a time of gift-giving. As fatiguing as 2020 and the COVID-19 pandemic have been, science has stepped up this year to provide humankind with a pair of truly hopeful gifts: the first two COVID-19 vaccines.
Two weeks ago, the U.S. Food and Drug Administration (FDA) granted emergency use authorization (EUA) to a COVID-19 vaccine from Pfizer/BioNTech, enabling distribution to begin to certain high-risk groups just three days later. More recently, the FDA granted an EUA to a COVID-19 vaccine from the biotechnology company Moderna, Cambridge, MA. This messenger RNA (mRNA) vaccine, which is part of a new approach to vaccination, was co-developed by NIH’s National Institute of Allergy and Infectious Diseases (NIAID). The EUA is based on data showing the vaccine is safe and 94.5 percent effective at protecting people from infection with SARS-CoV-2, the coronavirus that causes COVID-19.
Those data on the Moderna vaccine come from a clinical trial of 30,000 individuals, who generously participated to help others. We can’t thank those trial participants enough for this gift. The distribution of millions of Moderna vaccine doses is expected to begin this week.
It’s hard to put into words just how remarkable these accomplishments are in the history of science. A vaccine development process that used to take many years, often decades, has been condensed to about 11 months. Just last January, researchers started out with a previously unknown virus and we now have not just one, but two, vaccines that will be administered to millions of Americans before year’s end. And the accomplishments don’t end there—several other types of COVID-19 vaccines are also on the way.
It’s important to recognize that this couldn’t have happened without the efforts of many scientists working tirelessly behind the scenes for many years prior to the pandemic. Among those who deserve tremendous credit are Kizzmekia Corbett, Barney Graham, John Mascola, and other members of the amazing team at the Dale and Betty Bumpers Vaccine Research Center at NIH’s National Institute of Allergy and Infectious Diseases (NIAID).
When word of SARS-CoV-2 emerged, Corbett, Graham, and other NIAID researchers had already been studying other coronaviruses for years, including those responsible for earlier outbreaks of respiratory disease. So, when word came that this was a new coronavirus outbreak, they were ready to take action. It helped that they had paid special attention to the spike proteins on the surface of coronaviruses, which have turned out to be the main focus the COVID-19 vaccines now under development.
The two vaccines currently authorized for administration in the United States work in a unique way. Their centerpiece is a small, non-infectious snippet of mRNA. Our cells constantly produce thousands of mRNAs, which provide the instructions needed to make proteins. When someone receives an mRNA vaccine for COVID-19, it tells the person’s own cells to make the SARS-CoV-2 spike protein. The person’s immune system then recognizes the viral spike protein as foreign and produces antibodies to eliminate it.
This vaccine-spurred encounter trains the human immune system to remember the spike protein. So, if an actual SARS-CoV-2 virus tries to infect a vaccinated person weeks or months later, his or her immune system will be ready to fend it off. To produce the most vigorous and durable immunity against the virus, people will need to get two shots of mRNA vaccine, which are spaced several weeks to a month apart, depending on the vaccine.
Some have raised concerns on social media that mRNA vaccines might alter the DNA genome of someone being vaccinated. But that’s not possible, since this mRNA doesn’t enter the nucleus of the cell where DNA is located. Instead, the vaccine mRNAs stay in the outer part of the cell (the cytoplasm). What’s more, after being transcribed into protein just one time, the mRNA quickly degrades. Others have expressed concerns about whether the vaccine could cause COVID-19. That is not a risk because there’s no whole virus involved, just the coding instructions for the non-infectious spike protein.
An important advantage of mRNA is that it’s easy for researchers to synthesize once they know the nucleic acid sequence of a target viral protein. So, the gift of mRNA vaccines is one that will surely keep on giving. This new technology can now be used to speed the development of future vaccines. After the emergence of the disease-causing SARS, MERS, and now SARS-CoV-2 viruses, it would not be surprising if there are other coronavirus health threats in our future. Corbett and her colleagues are hoping to design a universal vaccine that can battle all of them. In addition, mRNA vaccines may prove effective for fighting future pandemics caused by other infectious agents and for preventing many other conditions, such as cancer and HIV.
Though vaccines are unquestionably our best hope for getting past the COVID-19 pandemic, public surveys indicate that some people are uneasy about accepting this disease-preventing gift. Some have even indicated they will refuse to take the vaccine. Healthy skepticism is a good thing, but decisions like this ought to be based on weighing the evidence of benefit versus risk. The results of the Pfizer and Moderna trials, all released for complete public scrutiny, indicate the potential benefits are high and the risks, low. Despite the impressive speed at which the new COVID-19 vaccines were developed, they have undergone and continue to undergo a rigorous process to generate all the data needed by the FDA to determine their long-term safety and effectiveness.
Unfortunately, the gift of COVID-19 vaccines comes too late for the more than 313,000 Americans who have died from complications of COVID-19, and many others who’ve had their lives disrupted and may have to contend with long-term health consequences related to COVID-19. The vaccines did arrive in record time, but all of us wish they could somehow have arrived even sooner to avert such widespread suffering and heartbreak.
It will be many months before all Americans who are willing to get a vaccine can be immunized. We need 75-80 percent of Americans to receive vaccines in order to attain the so-called “herd immunity” needed to drive SARS-CoV-2 away and allow us all to get back to a semblance of normal life.
Meanwhile, we all have a responsibility to do everything possible to block the ongoing transmission of this dangerous virus. Each of us needs to follow the three W’s: Wear a mask, Watch your distance, Wash your hands often.
When your chance for immunization comes, please roll up your sleeve and accept the potentially life-saving gift of a COVID-19 vaccine. In fact, I just got my first shot of the Moderna vaccine today along with NIAID Director Anthony Fauci, HHS Secretary Alex Azar, and some front-line healthcare workers at the NIH Clinical Center. Accepting this gift is our best chance to put this pandemic behind us, as we look forward to a better new year.
Links:
Coronavirus (COVID-19) (NIH)
Combat COVID (U.S. Department of Health and Human Services, Washington, D.C.)
Dale and Betty Bumpers Vaccine Research Center (National Institute of Allergy and Infectious Diseases/NIH)
Moderna (Cambridge, MA)
Pfizer (New York, NY)
BioNTech (Mainz, Germany)
New Online Resource Shows How You Can Help to Fight COVID-19
Posted on by Dr. Francis Collins

There are lots of useful online resources to learn about COVID-19 and some of the clinical studies taking place across the country. What’s been missing is a one-stop online information portal that pulls together the most current information for people of all groups, races, ethnicities, and backgrounds who want to get involved in fighting the pandemic. So, I’m happy to share that the U.S. Department of Health and Human Services, in coordination with NIH and Operation Warp Speed, has just launched a website called Combat COVID.
This easy-to-navigate portal makes it even easier for you and your loved ones to reach informed decisions about your health and to find out how to help in the fight against COVID-19. Indeed, it shows that no matter your current experience with COVID-19, there are opportunities to get involved to develop vaccines and medicines that will help everyone. Hundreds of thousands of volunteers have already taken this step—but we still need more, so we are seeking your help.
The Combat COVID website, which can also be viewed in Spanish, is organized to guide you to the most relevant information based on your own COVID-19 status:
• If you’ve never had COVID-19, you’ll be directed to information about joining the COVID-19 Prevention Network’s Volunteer Screening Registry. This registry is creating a list of potential volunteers willing to take part in ongoing or future NIH clinical trials focused on preventing COVID-19—like vaccines. Why get involved in a clinical trial now if vaccines will be widely distributed in the future? Well, there’s still a long way to go to get the pandemic under control, and several promising vaccines are still undergoing definitive testing. Your best route to getting access to a vaccine right now might be a clinical trial. And the more vaccines that are found to be safe and effective, the sooner we will be able to immunize all Americans and many others around the world.
• If you have an active COVID-19 infection, you’ll be directed to information about ongoing clinical trials that are studying better ways to treat the infection with promising drugs and other treatments. There are currently at least nine ongoing clinical trials for adults at every stage of COVID-19 illness. That includes five NIH Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) public-private partnership trials. All of these are promising treatments, but need to be rigorously tested to be sure they are safe and effective.
• If you’ve recovered from a confirmed case of COVID-19, you may be able to give the gift of life to someone else. Check out Combat COVID, where you’ll be directed to information about how to donate blood plasma. Once donated, this plasma may be infused into another person to help treat COVID-19 or it may be used to make a potential medicine.
• For doctors treating people with COVID-19, the website also provides a collection of useful information, including details on how to connect patients to ongoing clinical trials and other opportunities to combat COVID-19.
While I’m discussing online resources, NIH’s National Cancer Institute (NCI) also recently launched an interesting website for a critical initiative called the Serological Sciences Network for COVID-19 (SeroNet). A collaboration between several NIH components and 25 of the nation’s top biomedical research institutions, SeroNet will increase the national capacity for antibody testing, while also investigating all aspects of the immune response to SARS-CoV-2, the coronavirus that causes COVID-19. That includes studying variations in the severity of COVID-19 symptoms, the influence of pre-existing conditions for developing severe disease, and the chances of reinfection.
In our efforts to combat COVID-19, we’ve come a long way in a short period of time. But there is still plenty of work to do to get the pandemic under control to protect ourselves, our loved ones, and our communities. Be a hero. Follow the three W’s: Wear a mask. Watch your distance (stay 6 feet apart). Wash your hands often. And, if you’d like to find what else you can do to help, follow your way to Combat COVID.
Links:
Coronavirus (COVID-19) (NIH)
Combat COVID (U.S. Department of Health and Human Services, Washington, D.C.)
Explaining Operation Warp Speed (HHS)
Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) (NIH)
Serological Sciences Network for COVID-19 (SeroNet) (National Cancer Institute/NIH)
Next Page