clinical trials
Can Autoimmune Antibodies Explain Blood Clots in COVID-19?
Posted on by Dr. Francis Collins
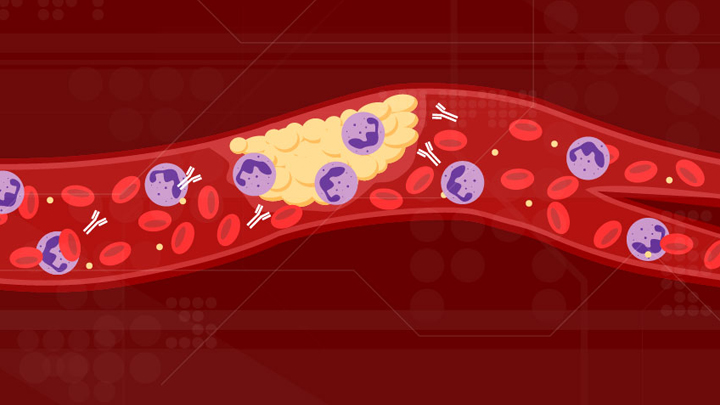
For people with severe COVID-19, one of the most troubling complications is abnormal blood clotting that puts them at risk of having a debilitating stroke or heart attack. A new study suggests that SARS-CoV-2, the coronavirus that causes COVID-19, doesn’t act alone in causing blood clots. The virus seems to unleash mysterious antibodies that mistakenly attack the body’s own cells to cause clots.
The NIH-supported study, published in Science Translational Medicine, uncovered at least one of these autoimmune antiphospholipid (aPL) antibodies in about half of blood samples taken from 172 patients hospitalized with COVID-19. Those with higher levels of the destructive autoantibodies also had other signs of trouble. They included greater numbers of sticky, clot-promoting platelets and NETs, webs of DNA and protein that immune cells called neutrophils spew to ensnare viruses during uncontrolled infections, but which can lead to inflammation and clotting. These observations, coupled with the results of lab and mouse studies, suggest that treatments to control those autoantibodies may hold promise for preventing the cascade of events that produce clots in people with COVID-19.
Our blood vessels normally strike a balance between producing clotting and anti-clotting factors. This balance keeps us ready to seal up vessels after injury, but otherwise to keep our blood flowing at just the right consistency so that neutrophils and platelets don’t stick and form clots at the wrong time. But previous studies have suggested that SARS-CoV-2 can tip the balance toward promoting clot formation, raising questions about which factors also get activated to further drive this dangerous imbalance.
To learn more, a team of physician-scientists, led by Yogendra Kanthi, a newly recruited Lasker Scholar at NIH’s National Heart, Lung, and Blood Institute and his University of Michigan colleague Jason S. Knight, looked to various types of aPL autoantibodies. These autoantibodies are a major focus in the Knight Lab’s studies of an acquired autoimmune clotting condition called antiphospholipid syndrome. In people with this syndrome, aPL autoantibodies attack phospholipids on the surface of cells including those that line blood vessels, leading to increased clotting. This syndrome is more common in people with other autoimmune or rheumatic conditions, such as lupus.
It’s also known that viral infections, including COVID-19, produce a transient increase in aPL antibodies. The researchers wondered whether those usually short-lived aPL antibodies in COVID-19 could trigger a condition similar to antiphospholipid syndrome.
The researchers showed that’s exactly the case. In lab studies, neutrophils from healthy people released twice as many NETs when cultured with autoantibodies from patients with COVID-19. That’s remarkably similar to what had been seen previously in such studies of the autoantibodies from patients with established antiphospholipid syndrome. Importantly, their studies in the lab further suggest that the drug dipyridamole, used for decades to prevent blood clots, may help to block that antibody-triggered release of NETs in COVID-19.
The researchers also used mouse models to confirm that autoantibodies from patients with COVID-19 actually led to blood clots. Again, those findings closely mirror what happens in mouse studies testing the effects of antibodies from patients with the most severe forms of antiphospholipid syndrome.
While more study is needed, the findings suggest that treatments directed at autoantibodies to limit the formation of NETs might improve outcomes for people severely ill with COVID-19. The researchers note that further study is needed to determine what triggers autoantibodies in the first place and how long they last in those who’ve recovered from COVID-19.
The researchers have already begun enrolling patients into a modest scale clinical trial to test the anti-clotting drug dipyridamole in patients who are hospitalized with COVID-19, to find out if it can protect against dangerous blood clots. These observations may also influence the design of the ACTIV-4 trial, which is testing various antithrombotic agents in outpatients, inpatients, and convalescent patients. Kanthi and Knight suggest it may also prove useful to test infected patients for aPL antibodies to help identify and improve treatment for those who may be at especially high risk for developing clots. The hope is this line of inquiry ultimately will lead to new approaches for avoiding this very troubling complication in patients with severe COVID-19.
Reference:
[1] Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Zuo Y, Estes SK, Ali RA, Gandhi AA, Yalavarthi S, Shi H, Sule G, Gockman K, Madison JA, Zuo M, Yadav V, Wang J, Woodard W, Lezak SP, Lugogo NL, Smith SA, Morrissey JH, Kanthi Y, Knight JS. Sci Transl Med. 2020 Nov 2:eabd3876.
Links:
Coronavirus (COVID-19) (NIH)
Antiphospholipid Antibody Syndrome (National Heart Lung and Blood Institute/NIH)
Kanthi Lab (National Heart, Lung, and Blood Institute, Bethesda, MD)
Knight Lab (University of Michigan)
ACTIV (NIH)
NIH Support: National Heart, Lung, and Blood Institute
Researchers Publish Encouraging Early Data on COVID-19 Vaccine
Posted on by Dr. Francis Collins

People all around the globe are anxiously awaiting development of a safe, effective vaccine to protect against the deadly threat of coronavirus disease 2019 (COVID-19). Evidence is growing that biomedical research is on track to provide such help, and to do so in record time.
Just two days ago, in a paper in the New England Journal of Medicine [1], researchers presented encouraging results from the vaccine that’s furthest along in U.S. human testing: an innovative approach from NIH’s Vaccine Research Center (VRC), in partnership with Moderna Inc., Cambridge, MA [1]. The centerpiece of this vaccine is a small, non-infectious snippet of messenger RNA (mRNA). Injecting this mRNA into muscle will spur a person’s own body to make a key viral protein, which, in turn, will encourage the production of protective antibodies against SARS-CoV-2—the novel coronavirus that causes COVID-19.
While it generally takes five to 10 years to develop a vaccine against a new infectious agent, we simply don’t have that time with a pandemic as devastating as COVID-19. Upon learning of the COVID-19 outbreak in China early this year, and seeing the genome sequence of SARS-CoV-2 appear on the internet, researchers with NIH’s National Institute of Allergy and Infectious Diseases (NIAID) carefully studied the viral instructions, focusing on the portion that codes for a spike protein that the virus uses to bind to and infect human cells.
Because of their experience with the original SARS virus back in the 2000s, they thought a similar approach to vaccine development would work and modified an existing design to reflect the different sequence of the SARS-CoV-2 spike protein. Literally within days, they had created a vaccine in the lab. They then went on to work with Moderna, a biotech firm that’s produced personalized cancer vaccines. All told, it took just 66 days from the time the genome sequence was made available in January to the start of the first-in-human study described in the new peer-reviewed paper.
In the NIH-supported phase 1 human clinical trial, researchers found the vaccine, called mRNA-1273, to be safe and generally well tolerated. Importantly, human volunteers also developed significant quantities of neutralizing antibodies that target the virus in the right place to block it from infecting their cells.
Conducted at Kaiser Permanente Washington Health Research Institute, Seattle; and Emory University School of Medicine, Atlanta, the trial led by Kaiser Permanente’s Lisa Jackson involved healthy adult volunteers. Each volunteer received two vaccinations in the upper arm at one of three doses, given approximately one month apart.
The volunteers will be tracked for a full year, allowing researchers to monitor their health and antibody production. However, the recently published paper provides interim data on the phase 1 trial’s first 45 participants, ages 18 to 55, for the first 57 days after their second vaccination. The data revealed:
• No volunteers suffered serious adverse events.
• Optimal dose to elicit high levels of neutralizing antibody activity, while also protecting patient safety, appears to be 100 micrograms. Doses administered in the phase 1 trial were either 25, 100, or 250 micrograms.
• More than half of the volunteers reported fatigue, headache, chills, muscle aches, or pain at the injection site. Those symptoms were most common after the second vaccination and in volunteers who received the highest vaccine dose. That dose will not be used in larger trials.
• Two doses of 100 micrograms of the vaccine prompted a robust immune response, which was last measured 43 days after the second dose. These responses were actually above the average levels seen in blood samples from people who had recovered from COVID-19.
These encouraging results are being used to inform the next rounds of human testing of the mRNA-1273 vaccine. A phase 2 clinical trial is already well on its way to recruiting 600 healthy adults.This study will continue to profile the vaccine’s safety, as well as its ability to trigger an immune response.
Meanwhile, later this month, a phase 3 clinical trial will begin enrolling 30,000 volunteers, with particular focus on recruitment in regions and populations that have been particularly hard hit by the virus.
The design of that trial, referred to as a “master protocol,” had major contributions from the Accelerating COVID-19 Therapeutic Interventions and Vaccine (ACTIV) initiative, a remarkable public-private partnership involving 20 biopharmaceutical companies, academic experts, and multiple federal agencies. Now, a coordinated effort across the U.S. government, called Operation Warp Speed, is supporting rapid conduct of these clinical trials and making sure that millions of doses of any successful vaccine will be ready if the vaccine proves save and effective.
Results of this first phase 3 trial are expected in a few months. If you are interested in volunteering for these or other prevention trials, please check out NIH’s new COVID-19 clinical trials network.
There’s still a lot of work that remains to be done, and anything can happen en route to the finish line. But by pulling together, and leaning on the very best science, I am confident that we will be able rise to the challenge of ending this pandemic that has devastated so many lives.
Reference:
[1] A SARS-CoV-2 mRNA Vaccine—Preliminary Report. Jackson LA, Anderson EJ, Rouphael NG, Ledgerwood JE, Graham BS, Beigel JH, et al. NEJM. 2020 July 14. [Publication ahead of print]
Links:
Coronavirus (COVID-19) (NIH)
Dale and Betty Bumpers Vaccine Research Center (National Institute of Allergy and Infectious Diseases/NIH)
Moderna, Inc. (Cambridge, MA)
Safety and Immunogenicity Study of 2019-nCoV Vaccine (mRNA-1273) for Prophylaxis of SARS-CoV-2 Infection (COVID-19) (ClinicalTrials.gov)
“NIH Launches Clinical Trials Network to Test COVID-19 Vaccines and Other Prevention Tools,” NIAID News Release, NIH, July 8, 2020.
Accelerating COVID-19 Therapeutic Interventions and Vaccines (ACTIV) (NIH)
Explaining Operation Warp Speed (U.S. Department of Health and Human Services, Washington, DC)
NIH Support: National Institute of Allergy and Infectious Diseases
Pursuing Safe and Effective Anti-Viral Drugs for COVID-19
Posted on by Dr. Francis Collins

Right now, the world is utterly focused on the coronavirus outbreak known as COVID-19. That’s certainly true for those of us at NIH. Though I am working from home to adhere rigorously to physical distancing, I can’t remember ever working harder, trying to do everything I can to assist in the development of safe and effective treatments and vaccines.
Over the past several weeks, a mind-boggling array of possible therapies have been considered. None have yet been proven to be effective in rigorously controlled trials, but for one of them, it’s been a busy week. So let’s focus on an experimental anti-viral drug, called remdesivir, that was originally developed for the deadly Ebola virus. Though remdesivir failed to help people with Ebola virus disease, encouraging results from studies of coronavirus-infected animals have prompted the launch of human clinical trials to see if this drug might fight SARS-CoV-2, the novel coronavirus that causes COVID-19.
You may wonder how a drug could possibly work for Ebola and SARS-CoV-2, since they are very different viruses that produce dramatically different symptoms in humans. The commonality is that both viruses have genomes made of ribonucleic acid (RNA), which must be copied by an enzyme called RNA-dependent RNA polymerase for the virus to replicate.
Remdesivir has an affinity for attaching to this kind of polymerase because its structure is very similar to one of the RNA letters that make up the viral genome [1]. Due to this similarity, when an RNA virus attempts to replicate, its polymerase is tricked into incorporating remdesivir into its genome as a foreign nucleotide, or anomalous letter. That undecipherable, extra letter brings the replication process to a crashing halt—and, without the ability to replicate, viruses can’t infect human cells.
Would this work on a SARS-CoV-2 infection in a living organism? An important step was just posted as a preprint yesterday—a small study showed infusion of remdesivir was effective in limiting the severity of lung disease in rhesus macaques [2]. That’s encouraging news. But the only sure way to find out if remdesivir will actually help humans who are infected with SARS-CoV-2 is to conduct a randomized, controlled clinical trial.
In late February, NIH’s National Institute of Allergy and Infectious Diseases (NIAID) did just that, when it launched a randomized, controlled clinical trial to test remdesivir in people with COVID-19. The study, led by NIAID’s Division of Microbiology and Infectious Diseases, has already enrolled 805 patients at 67 testing sites. Most sites are in the United States, but there are also some in Singapore, Japan, South Korea, Mexico, Spain, the United Kingdom, Denmark, Greece, and Germany.
All trial participants must have laboratory-confirmed COVID-19 infections and evidence of lung involvement, such as abnormal chest X-rays, rattling sounds when breathing (rales) with a need for supplemental oxygen, or a need for mechanical ventilation. They are randomly assigned to receive either a round of treatment with remdesivir or a harmless placebo with no therapeutic effect. To avoid bias from creeping into patient care, the study is double-blind, meaning neither the medical staff nor the participants know who is receiving remdesivir.
There is also an early hint from another publication that remdesivir may benefit some people with COVID-19. Since the end of January 2020, Gilead Sciences, Foster City, CA, which makes remdesivir, has provided daily, intravenous infusions of the drug on a compassionate basis to more than 1,800 people hospitalized with advanced COVID-19 around the world. In a study of a subgroup of 53 compassionate-use patients with advanced complications of COVID-19, nearly two-thirds improved when given remdesivir for up to 10 days [3]. Most of the participants were men over age 60 with preexisting conditions that included hypertension, diabetes, high cholesterol, and asthma.
This may sound exciting, but these preliminary results, published in the New England Journal of Medicine, come with major caveats. There were no controls, participants were not randomized, and the study lacked other key features of the more rigorously designed NIH clinical trial. We can all look forward to the results from the NIH trial, which are are expected within a matter of weeks. Hopefully these will provide much-needed scientific evidence on remdesivir’s safety and efficacy in people with COVID-19.
In the meantime, basic researchers continue to learn more about remdesivir and its interaction with the novel coronavirus that causes COVID-19. In a recent study in the journal Science, a research team, led by Quan Wang, Shanghai Tech University, China, mapped the 3D atomic structure of the novel coronavirus’s polymerase while it was complexed with two other vital parts of the viral replication machinery [4]. This was accomplished using a high-resolution imaging approach called cryo-electron microscopy (cryo-EM), which involves flash-freezing molecules in liquid nitrogen and bombarding them with electrons to capture their images with a special camera.
With these atomic structures in hand, the researchers then modeled exactly how remdesivir binds to the polymerase of the novel coronavirus. The model will help inform future efforts to tweak the structure of the drug and optimize its ability to disrupt viral replication. Such detailed biochemical information will be vital in the weeks ahead, especially if data generated by the NIH clinical trial indicate that remdesivir is a worthwhile lead to pursue in our ongoing search for anti-viral drugs to combat the global COVID-19 pandemic.
References:
[1] Nucleoside analogues for the treatment of coronavirus infections. Pruijssers AJ, Denison MR. Curr Opin Virol. 2019 Apr;35:57-62.
[2] Clinical benefit of remdesivir in rhesus macaques infected with SARS-CoV-2. Williamson B, Feldmann F, Schwarz B, Scott D, Munster V, de Wit E et. al. BioRxiv. Preprint posted 15 April 2020.
[3] Compassionate use of remdesivir for patients with severe Covid-19. Grein J, Ohmagari N, Shin D, Brainard DM, Childs R, Flanigan T. et. al. N Engl J Med. 2020 Apr 10. [Epub ahead of publication]
[4] Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Gao Y, Yan L, Liu F, Wang Q, Lou Z, Rao A, et al. Science. 10 April 2020. [Epub ahead of publication]
Links:
Coronavirus (COVID-19) (NIH)
Accelerating COVID-19 Therapeutic Interventions and Vaccines (NIH)
NIH Clinical Trial of Remdesivir to Treat COVID-19 Begins (National Institute of Allergy and Infectious Diseases/NIH)
Developing Therapeutics and Vaccines for Coronaviruses (NIAID)
COVID-19, MERS & SARS (NIAID)
NIH Support: National Institute of Allergy and Infectious Diseases
Structural Biology Points Way to Coronavirus Vaccine
Posted on by Dr. Francis Collins
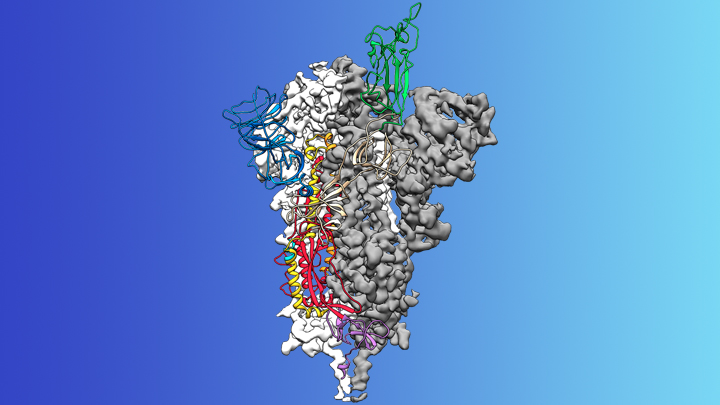
Credit: McLellan Lab, University of Texas at Austin
The recent COVID-19 outbreak of a novel type of coronavirus that began in China has prompted a massive global effort to contain and slow its spread. Despite those efforts, over the last month the virus has begun circulating outside of China in multiple countries and territories.
Cases have now appeared in the United States involving some affected individuals who haven’t traveled recently outside the country. They also have had no known contact with others who have recently arrived from China or other countries where the virus is spreading. The NIH and other U.S. public health agencies stand on high alert and have mobilized needed resources to help not only in its containment, but in the development of life-saving interventions.
On the treatment and prevention front, some encouraging news was recently reported. In record time, an NIH-funded team of researchers has created the first atomic-scale map of a promising protein target for vaccine development [1]. This is the so-called spike protein on the new coronavirus that causes COVID-19. As shown above, a portion of this spiky surface appendage (green) allows the virus to bind a receptor on human cells, causing other portions of the spike to fuse the viral and human cell membranes. This process is needed for the virus to gain entry into cells and infect them.
Preclinical studies in mice of a candidate vaccine based on this spike protein are already underway at NIH’s Vaccine Research Center (VRC), part of the National Institute of Allergy and Infectious Diseases (NIAID). An early-stage phase I clinical trial of this vaccine in people is expected to begin within weeks. But there will be many more steps after that to test safety and efficacy, and then to scale up to produce millions of doses. Even though this timetable will potentially break all previous speed records, a safe and effective vaccine will take at least another year to be ready for widespread deployment.
Coronaviruses are a large family of viruses, including some that cause “the common cold” in healthy humans. In fact, these viruses are found throughout the world and account for up to 30 percent of upper respiratory tract infections in adults.
This outbreak of COVID-19 marks the third time in recent years that a coronavirus has emerged to cause severe disease and death in some people. Earlier coronavirus outbreaks included SARS (severe acute respiratory syndrome), which emerged in late 2002 and disappeared two years later, and MERS (Middle East respiratory syndrome), which emerged in 2012 and continues to affect people in small numbers.
Soon after COVID-19 emerged, the new coronavirus, which is closely related to SARS, was recognized as its cause. NIH-funded researchers including Jason McLellan, an alumnus of the VRC and now at The University of Texas at Austin, were ready. They’d been studying coronaviruses in collaboration with NIAID investigators for years, with special attention to the spike proteins.
Just two weeks after Chinese scientists reported the first genome sequence of the virus [2], McLellan and his colleagues designed and produced samples of its spike protein. Importantly, his team had earlier developed a method to lock coronavirus spike proteins into a shape that makes them both easier to analyze structurally via the high-resolution imaging tool cryo-electron microscopy and to use in vaccine development efforts.
After locking the spike protein in the shape it takes before fusing with a human cell to infect it, the researchers reconstructed its atomic-scale 3D structural map in just 12 days. Their results, published in Science, confirm that the spike protein on the virus that causes COVID-19 is quite similar to that of its close relative, the SARS virus. It also appears to bind human cells more tightly than the SARS virus, which may help to explain why the new coronavirus appears to spread more easily from person to person, mainly by respiratory transmission.
McLellan’s team and his NIAID VRC counterparts also plan to use the stabilized spike protein as a probe to isolate naturally produced antibodies from people who’ve recovered from COVID-19. Such antibodies might form the basis of a treatment for people who’ve been exposed to the virus, such as health care workers.
The NIAID is now working with the biotechnology company Moderna, Cambridge, MA, to use the latest findings to develop a vaccine candidate using messenger RNA (mRNA), molecules that serve as templates for making proteins. The goal is to direct the body to produce a spike protein in such a way to elicit an immune response and the production of antibodies. An early clinical trial of the vaccine in people is expected to begin in the coming weeks. Other vaccine candidates are also in preclinical development.
Meanwhile, the first clinical trial in the U.S. to evaluate an experimental treatment for COVID-19 is already underway at the University of Nebraska Medical Center’s biocontainment unit [3]. The NIH-sponsored trial will evaluate the safety and efficacy of the experimental antiviral drug remdesivir in hospitalized adults diagnosed with COVID-19. The first participant is an American who was repatriated after being quarantined on the Diamond Princess cruise ship in Japan.
As noted, the risk of contracting COVID-19 in the United States is currently low, but the situation is changing rapidly. One of the features that makes the virus so challenging to stay in front of is its long latency period before the characteristic flu-like fever, cough, and shortness of breath manifest. In fact, people infected with the virus may not show any symptoms for up to two weeks, allowing them to pass it on to others in the meantime. You can track the reported cases in the United States on the Centers for Disease Control and Prevention’s website.
As the outbreak continues over the coming weeks and months, you can be certain that NIH and other U.S. public health organizations are working at full speed to understand this virus and to develop better diagnostics, treatments, and vaccines.
References:
[1] Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS. Science. 2020 Feb 19.
[2] A new coronavirus associated with human respiratory disease in China. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, Zhang YZ. Nature. 2020 Feb 3.
[3] NIH clinical trial of remdesivir to treat COVID-19 begins. NIH News Release. Feb 25, 2020.
Links:
Coronaviruses (National Institute of Allergy and Infectious Diseases/NIH)
Coronavirus (COVID-19) (NIAID)
Coronavirus Disease 2019 (Centers for Disease Control and Prevention, Atlanta)
NIH Support: National Institute of Allergy and Infectious Diseases
Celebrating 2019 Biomedical Breakthroughs
Posted on by Dr. Francis Collins

Happy New Year! As we say goodbye to the Teens, let’s take a look back at 2019 and some of the groundbreaking scientific discoveries that closed out this remarkable decade.
Each December, the reporters and editors at the journal Science select their breakthrough of the year, and the choice for 2019 is nothing less than spectacular: An international network of radio astronomers published the first image of a black hole, the long-theorized cosmic singularity where gravity is so strong that even light cannot escape [1]. This one resides in a galaxy 53 million light-years from Earth! (A light-year equals about 6 trillion miles.)
Though the competition was certainly stiff in 2019, the biomedical sciences were well represented among Science’s “runner-up” breakthroughs. They include three breakthroughs that have received NIH support. Let’s take a look at them:
In a first, drug treats most cases of cystic fibrosis: Last October, two international research teams reported the results from phase 3 clinical trials of the triple drug therapy Trikafta to treat cystic fibrosis (CF). Their data showed Trikafta effectively compensates for the effects of a mutation carried by about 90 percent of people born with CF. Upon reviewing these impressive data, the Food and Drug Administration (FDA) approved Trikafta, developed by Vertex Pharmaceuticals.
The approval of Trikafta was a wonderful day for me personally, having co-led the team that isolated the CF gene 30 years ago. A few years later, I wrote a song called “Dare to Dream” imagining that wonderful day when “the story of CF is history.” Though we’ve still got more work to do, we’re getting a lot closer to making that dream come true. Indeed, with the approval of Trikafta, most people with CF have for the first time ever a real chance at managing this genetic disease as a chronic condition over the course of their lives. That’s a tremendous accomplishment considering that few with CF lived beyond their teens as recently as the 1980s.
Such progress has been made possible by decades of work involving a vast number of researchers, many funded by NIH, as well as by more than two decades of visionary and collaborative efforts between the Cystic Fibrosis Foundation and Aurora Biosciences (now, Vertex) that built upon that fundamental knowledge of the responsible gene and its protein product. Not only did this innovative approach serve to accelerate the development of therapies for CF, it established a model that may inform efforts to develop therapies for other rare genetic diseases.
Hope for Ebola patients, at last: It was just six years ago that news of a major Ebola outbreak in West Africa sounded a global health emergency of the highest order. Ebola virus disease was then recognized as an untreatable, rapidly fatal illness for the majority of those who contracted it. Though international control efforts ultimately contained the spread of the virus in West Africa within about two years, over 28,600 cases had been confirmed leading to more than 11,000 deaths—marking the largest known Ebola outbreak in human history. Most recently, another major outbreak continues to wreak havoc in northeastern Democratic Republic of Congo (DRC), where violent civil unrest is greatly challenging public health control efforts.
As troubling as this news remains, 2019 brought a needed breakthrough for the millions of people living in areas susceptible to Ebola outbreaks. A randomized clinical trial in the DRC evaluated four different drugs for treating acutely infected individuals, including an antibody against the virus called mAb114, and a cocktail of anti-Ebola antibodies referred to as REGN-EB3. The trial’s preliminary data showed that about 70 percent of the patients who received either mAb114 or the REGN-EB3 antibody cocktail survived, compared with about half of those given either of the other two medicines.
So compelling were these preliminary results that the trial, co-sponsored by NIH’s National Institute of Allergy and Infectious Diseases (NIAID) and the DRC’s National Institute for Biomedical Research, was halted last August. The results were also promptly made public to help save lives and stem the latest outbreak. All Ebola patients in the DRC treatment centers now are treated with one or the other of these two options. The trial results were recently published.
The NIH-developed mAb114 antibody and the REGN-EB3 cocktail are the first therapeutics to be shown in a scientifically rigorous study to be effective at treating Ebola. This work also demonstrates that ethically sound clinical research can be conducted under difficult conditions in the midst of a disease outbreak. In fact, the halted study was named Pamoja Tulinde Maisha (PALM), which means “together save lives” in Kiswahili.
To top off the life-saving progress in 2019, the FDA just approved the first vaccine for Ebola. Called Ervebo (earlier rVSV-ZEBOV), this single-dose injectable vaccine is a non-infectious version of an animal virus that has been genetically engineered to carry a segment of a gene from the Zaire species of the Ebola virus—the virus responsible for the current DRC outbreak and the West Africa outbreak. Because the vaccine does not contain the whole Zaire virus, it can’t cause Ebola. Results from a large study in Guinea conducted by the WHO indicated that the vaccine offered substantial protection against Ebola virus disease. Ervebo, produced by Merck, has already been given to over 259,000 individuals as part of the response to the DRC outbreak. The NIH has supported numerous clinical trials of the vaccine, including an ongoing study in West Africa.
Microbes combat malnourishment: Researchers discovered a few years ago that abnormal microbial communities, or microbiomes, in the intestine appear to contribute to childhood malnutrition. An NIH-supported research team followed up on this lead with a study of kids in Bangladesh, and it published last July its groundbreaking finding: that foods formulated to repair the “gut microbiome” helped malnourished kids rebuild their health. The researchers were able to identify a network of 15 bacterial species that consistently interact in the gut microbiomes of Bangladeshi children. In this month-long study, this bacterial network helped the researchers characterize a child’s microbiome and/or its relative state of repair.
But a month isn’t long enough to determine how the new foods would help children grow and recover. The researchers are conducting a similar study that is much longer and larger. Globally, malnutrition affects an estimated 238 million children under the age 5, stunting their normal growth, compromising their health, and limiting their mental development. The hope is that these new foods and others adapted for use around the world soon will help many more kids grow up to be healthy adults.
Measles Resurgent: The staff at Science also listed their less-encouraging 2019 Breakdowns of the Year, and unfortunately the biomedical sciences made the cut with the return of measles in the U.S. Prior to 1963, when the measles vaccine was developed, 3 to 4 million Americans were sickened by measles each year. Each year about 500 children would die from measles, and many more would suffer lifelong complications. As more people were vaccinated, the incidence of measles plummeted. By the year 2000, the disease was even declared eliminated from the U.S.
But, as more parents have chosen not to vaccinate their children, driven by the now debunked claim that vaccines are connected to autism, measles has made a very preventable comeback. Last October, the Centers for Disease Control and Prevention (CDC) reported an estimated 1,250 measles cases in the United States at that point in 2019, surpassing the total number of cases reported annually in each of the past 25 years.
The good news is those numbers can be reduced if more people get the vaccine, which has been shown repeatedly in many large and rigorous studies to be safe and effective. The CDC recommends that children should receive their first dose by 12 to 15 months of age and a second dose between the ages of 4 and 6. Older people who’ve been vaccinated or have had the measles previously should consider being re-vaccinated, especially if they live in places with low vaccination rates or will be traveling to countries where measles are endemic.
Despite this public health breakdown, 2019 closed out a memorable decade of scientific discovery. The Twenties will build on discoveries made during the Teens and bring us even closer to an era of precision medicine to improve the lives of millions of Americans. So, onward to 2020—and happy New Year!
Reference:
[1] 2019 Breakthrough of the Year. Science, December 19, 2019.
NIH Support: These breakthroughs represent the culmination of years of research involving many investigators and the support of multiple NIH institutes.
Dare to Dream: The Long Road to Targeted Therapies for Cystic Fibrosis
Posted on by Dr. Francis Collins

When your world has been touched by a life-threatening disease, it’s hard to spend a lot of time dreaming about the future. But that’s exactly what Jenny, an 8-year-old girl with cystic fibrosis (CF), did 30 years ago upon hearing the news that I and my colleagues in Ann Arbor and Toronto had discovered the gene for CF [1,2]. Her upbeat diary entry, which you can read above, is among the many ways in which people with CF have encouraged researchers on the long and difficult road toward achieving our shared dream of effective, molecularly targeted therapies for one of the nation’s most common potentially fatal recessive genetic diseases, affecting more than 30,000 individuals in the United States [3].
Today, I’m overjoyed to say that this dream finally appears to have come true for about 90 percent of people with CF. In papers in the New England Journal of Medicine and The Lancet [4,5], two international teams, including researchers partly supported by NIH, report impressive results from phase 3 clinical trials of a triple drug therapy for individuals with CF and at least one copy of Phe508del, the most common CF-causing mutation. And Jenny happens to be among those who now stand to benefit from this major advance.
Now happily married and living in Colorado, Jenny is leading an active life, writing a children’s book and trying to keep up with her daughter Pippa Lou, whom you see with her in the photo above. In a recent email to me, her optimistic outlook continues to shine through: “I have ALWAYS known in my heart that CF will be cured during my lifetime and I have made it my goal to be strong and ready for that day when it comes. None of the advancements in care would be what they are without you.”
But there are a great many more people who need to be recognized and thanked. Such advances were made possible by decades of work involving a vast number of other researchers, many funded by NIH, as well as by more than two decades of visionary and collaborative efforts between the Cystic Fibrosis Foundation and Aurora Biosciences (now, Vertex Pharmaceuticals) that built upon that fundamental knowledge of the responsible gene and its protein product. Not only did this innovative approach serve to accelerate the development of therapies for CF, it established a model that may inform efforts to develop therapies for other rare genetic diseases.
To understand how the new triple therapy works, one first needs to understand some things about the protein affected by CF, the cystic fibrosis transmembrane regulator (CFTR). In healthy people, CFTR serves as a gated channel for chloride ions in the cell membrane, regulating the balance of salt and water in the lungs, pancreas, sweat glands, and other organ systems.
People with the most common CF-causing Phe508del mutation produce a CFTR protein with two serious problems: misfolding that often results in the protein becoming trapped in the cell’s factory production line called the endoplasmic reticulum; and deficient activation of any protein that does manage to reach its proper location in the cell membrane. Consequently, an effective therapy for such people needs to include drugs that can correct the CFTR misfolding, along with those than can activate, or potentiate, the function of CFTR when it reaches the cell membrane.
The new triple combination therapy, which was developed by Vertex Pharmaceuticals and recently approved by the Food and Drug Administration (FDA) [6], is elexacaftor-tezacaftor-ivacaftor (two correctors and one potentiator). This approach builds upon the success of ivacaftor monotherapy, approved by the FDA in 2012 for rare CF-causing mutations; and tezacaftor-ivacaftor dual therapy, approved by the FDA in 2018 for people with two copies of the Phe508del mutation.
Specifically, the final results from two Phase 3 multi-center, randomized clinical trials demonstrated the safety and efficacy of the triple combination therapy for people with either one or two copies of the Phe508del mutation—which represents about 90 percent of people with CF. Patients in both trials experienced striking improvements in a key measure of lung capacity (forced expiratory volume in 1 second) and in sweat chloride levels, which show if the drugs are working throughout the body. In addition, the triple therapy was generally safe and well tolerated, with less than 1 percent of patients discontinuing the treatment due to adverse effects.
This is wonderful news! But let’s be clear—we are not yet at our journey’s end when it comes to realizing the full dream of defeating CF. More work remains to be done to help the approximately 10 percent of CF patients whose mutations result in the production of virtually no CFTR protein, which means there is nothing for current drugs to correct or activate.
Beyond that, wouldn’t it be great if biomedical science could figure out a way to permanently cure CF, perhaps using nonheritable gene editing, so no one needs to take drugs at all? It’s a bold dream, but look how far a little dreaming, plus a lot of hard work, has taken us so far in Jenny’s life.
In closing, I’d like to leave you with the chorus of a song, called “Dare to Dream,” that I wrote shortly after we identified the CF gene. I hope the words inspire not only folks affected by CF, but everyone who is looking to NIH-supported research for healing and hope.
Dare to dream, dare to dream,
All our brothers and sisters breathing free.
Unafraid, our hearts unswayed,
‘Til the story of CF is history.
References:
[1]. Identification of the cystic fibrosis gene: chromosome walking and jumping. Rommens JM, Iannuzzi MC, Kerem B, et al. Science 1989; 245:1059-1065.
[2]. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Riordan JR, Rommens JM, Kerem B, et al. Science 1989; 245:1066-73. Erratum in: Science 1989; 245:1437.
[3] Realizing the Dream of Molecularly Targeted Therapies for Cystic Fibrosis. Collins, FS. N Engl J Med. 2019 Oct 31. [Epub ahead of print]
[4]. Elexacaftor-Tezacaftor-Ivacaftor for CF with a Single Phe508del Mutation. Middleton P, Mall M, Drevinek P, et al.N Engl J Med. 2019 Oct 31. [Epub ahead of print]
[5] Efficacy and safety of the elexacaftor/tezacaftor/ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. Heijerman H, McKone E, Downey D, et al. Lancet. 2019 Oct 31. [Epub ahead of print]
[6] FDA approves new breakthrough therapy for cystic fibrosis. FDA News Release, Oct. 21, 2019.
Links:
Cystic Fibrosis (Genetics Home Reference/National Library of Medicine/NIH)
Research Milestones (Cystic Fibrosis Foundation, Bethesda, MD)
Wheezie Stevens in “Bubbles Can’t Hold Rain,” by Jennifer K. McGlincy
NIH Support: National, Heart, Lung and Blood Institute; National Institute of Diabetes and Digestive and Kidney Diseases; National Center for Advancing Translational Sciences
In Memory of Andrew Lee
Posted on by Dr. Francis Collins

A lot of young people are driven—driven to get a good education, land a great job, find true love, or see the world. But, today, I want to honor the life of a young man who was driven by something even bigger. Andrew Lee was driven to cure kidney cancer—not only for himself, but for others as well.
I knew and loved Andrew. And so did the legion of doctors, nurses, researchers, and other team members who had the privilege of fighting cancer with him over four very challenging years. Andrew was 19, just finishing his freshman year of college, when he received a devastating diagnosis: stage 4 kidney cancer, a rare type called Hereditary Leiomyomatosis and Renal Cell Cancer (HLRCC). There is no known cure for HLRCC, and doctors estimated his survival at about a year at best.
Still, Andrew and his family weren’t about to go hide somewhere and wait for the end. They began a journey that led him to take part in at least seven clinical trials, including ones at Yale University, New Haven, CT; Georgetown University, Washington, DC; and the NIH Clinical Center, Bethesda, MD. Experimental treatments slowed down the cancer, but sometimes made him terribly sick. Yet, Andrew always remained optimistic and cheerful. If a trial didn’t help him, maybe it would help someone else.
Andrew’s generosity didn’t stop there. Inspired by his father’s gift of a totally awesome 2015 Liberty Walk Nissan GT-R, he founded the Driven To Cure (DTC) nonprofit and traveled the country in his orange sports car to raise funds for kidney cancer research. According to the National Cancer Institute, nearly 63,000 Americans are diagnosed with kidney and renal pelvis cancers each year.
Andrew figured out how to put the “fun” in fundraising, drawing crowds at car shows and raising more than $500,000 in donations in just three years. His efforts were recognized by the Foundation for the NIH’s Charlie Sanders Award, which I had the privilege of presenting to him last fall.
But I think it was Andrew’s humanity that touched us the most. He always had time to share his story, to encourage another child or adult struggling with a frightening diagnosis. He’d give thrills to kids at The Children’s Inn at NIH when he rumbled into the parking lot with his 700 horsepower GT-R. At car shows, throngs of people were drawn in by the turbocharged ride and then captivated by the young man with the bright smile and compelling story. Andrew wrote: “I realized that the vehicle of my dreams was also the vehicle which gave me the opportunity to make a difference; to do something bigger than myself.”
Still, on the personal level, kidney cancer proved relentless. Options for treatment eventually ran out. As the disease progressed, Andrew and his family had to make another difficult transition—choosing to celebrate life, even in the face of its approaching end. He needed a wheelchair, so family and friends came up with one, keeping in mind one of Andrew’s last wishes. When Andrew needed 24-hour care and pain control, he was admitted to the NIH Clinical Center Hospice Unit, where comfort could be provided and his loved ones could gather around. That even included getting government permission for a visit from his dog Milo! Surrounded by friends and family, he died peacefully on April 21.
Andrew made friends with everyone—especially kids at The Children’s Inn. One special buddy was Isaac Barchus, who has a rare autoinflammatory disease called CANDLE Syndrome. When he was back home in Omaha, NE, Isaac enjoyed challenging Andrew to long-distance video games, especially FIFA Soccer.
Although Isaac can walk, it can be very painful, so he sometimes turned to an old, beat-up wheelchair to cover long distances. But not anymore. When Isaac turned 15 on June 7, Andrew’s father Bruce Lee fulfilled his son’s wish for the future of his wheelchair. He presented Isaac with Andrew’s wheelchair, which had now been painted the same orange color as Andrew’s GT-R and emblazoned with the feisty slogan on Andrew’s personalized license plate—F CANCR. What a cool birthday gift!
During his final weeks and days, Andrew and his dad often listened to the Andy Grammer song, “Don’t Give Up on Me.” Andrew’s family never gave up on him, and he never gave up on them either. In fact, Andrew never gave up caring, loving, and believing. He wouldn’t want us to either, as his favorite song reminds us: “I will fight, I will fight for you; I always do until my heart is black and blue.”
Yes, Andrew, our hearts are black and blue from losing you. But we won’t give up on all you stood for in your short but inspiring life. Race In Peace, dear Andrew.
Links:
Remembering Andrew Lee (Foundation for the National Institutes of Health)
NIH Cancer Patient Receives Humanitarian Award (The NIH Record)
Driven To Cure (Silver Spring, MD)
Video: Fighting Cancer With a 700-hp Nissan GT-R (The Drive)
Video: Andy Grammer—”Don’t Give Up On Me” [Official Lyric Video] from the film Five Feet Apart
Hereditary Leiomyomatosis and Renal Cell Cancer (National Library of Medicine/NIH)
Kidney (Renal Cell) Cancer (National Cancer Institute/NIH)
CANDLE Syndrome (Genetic and Rare Diseases Information Center/NIH)
Treating CANDLE Syndrome (National Institute of Allergy and Infectious Diseases/NIH)
Accelerating Cures in the Genomic Age: The Sickle Cell Example
Posted on by Dr. Francis Collins

Forty-five years ago, when I was a first-year medical student, a lecturer introduced me to a young man with sickle cell disease (SCD). Sickle cell disease is the first “molecular disease”, with its cause having been identified decades ago. That helped me see the connection between the abstract concepts of molecular genetics and their real-world human consequences in a way no textbook could. In fact, it inspired some of my earliest research on human hemoglobin disorders, which I conducted as a postdoctoral fellow.
Today, I’m heartened to report that, thanks to decades of biomedical advances, we stand on the verge of a cure for SCD. While at the American Society of Hematology meeting in San Diego last week, I was excited to be part of a discussion about how the tools and technologies arising from the Human Genome Project are accelerating the quest for cures.
The good news at the meeting included some promising, early results from human clinical trials of SCD gene therapies, including new data from the NIH Clinical Center. Researchers also presented very encouraging pre-clinical work on how gene-editing technologies, such as CRISPR, can be used in ways that may open the door to curing everyone with SCD. In fact, just days before the meeting, the first clinical trial for a CRISPR approach to SCD opened.
One important note: the gene editing research aimed at curing SCD is being done in non-reproductive (somatic) cells. The NIH does not support the use of gene editing technologies in human embryos (germline). I recently reiterated our opposition to germline gene editing, in response to an unethical experiment by a researcher in China who claims to have used CRISPR editing on embryos to produce twin girls resistant to HIV.
SCD affects approximately 100,000 people in the United States, and another 20 million worldwide, mostly in developing nations. This inherited, potentially life-threatening disorder is caused by a specific point mutation in a gene that codes for the beta chain of hemoglobin, a molecule found in red blood cells that deliver oxygen throughout the body. In people with SCD, the mutant hemoglobin forms insoluble aggregates when de-oxygenated. As a result the red cells assume a sickle shape, rather than the usual donut shape. These sickled cells clump together and stick in small blood vessels, resulting in severe pain, blood cell destruction, anemia, stroke, pulmonary hypertension, organ failure, and much too often, early death.
The need for a widespread cure for SCD is great. Since 1998, doctors have used a drug called hydroxyurea to reduce symptoms, but it can cause serious side effects and increase the risk of certain cancers. Blood transfusions can also ease symptoms in certain instances, but they too come with risks and complications. At the present time, the only way to cure SCD is a bone marrow transplant. However, transplants are not an option for many patients due to lack of matched marrow donors.
The good news is that novel genetic approaches have raised hopes of a widespread cure for SCD, possibly even within five to 10 years. So, in September, NIH’s National Heart, Lung, and Blood Institute launched the Cure Sickle Cell Initiative to accelerate development of the most promising of these next generation of therapies
At the ASH meeting, that first wave of this progress was evident. A team led by NHLBI’s John Tisdale, in collaboration with Bluebird bio, Cambridge, MA, was among the groups that presented impressive early results from human clinical trials testing novel gene replacement therapies for SCD. In the NIH trial, researchers removed blood precursor cells, called hematopoietic stem cells (HSCs), from a patient’s own bone marrow or bloodstream and used a harmless virus to insert a sickle-resistant hemoglobin gene. Then, after a chemotherapy infusion to condition the patient’s existing bone marrow, they returned the corrected cells to the patient.
So far, nine SCD patients have received the most advanced form of the experimental gene therapy, and Tisdale presented data on those who were farthest out from treatment [1,2]. His team found that in the four patients who were at least six months out, levels of gene therapy-derived hemoglobin were found to equal or exceed their levels of SCD hemoglobin.
Very cool science, but what does this mean for SCD patients’ health and well-being? Well, none of the gene therapy trial participants have required a blood transfusion during the follow-up period. In addition, improvements were seen in their hemoglobin levels and key markers of blood-cell destruction (total bilirubin concentration, lactate dehydrogenase, and reticulocyte counts) compared to baseline. Most importantly, in the years leading up to the clinical trial, all of the participants had experienced frequent painful sickle crises, in which sickled cells blocked their blood vessels. No such episodes were reported among the participants in the months after they received the gene therapy.
Researchers did report that one patient receiving this form of gene therapy developed myelodysplastic syndrome (MDS), a serious condition in which the blood-forming cells in the bone marrow become abnormal. However, there is no indication that the gene replacement technology itself caused the problem, and MDS has previously been linked to the chemotherapy drugs used in conditioning regimens before bone marrow transplants.
The NIH trial is just one of several clinical trials for SCD that are using viral vectors to deliver a variety of genes with therapeutic potential. Other trials actively recruiting are led by researchers at Boston Children’s Hospital, Cincinnati Children’s Hospital, and the University of California, Los Angeles.
While it’s hoped that genes inserted by viral vectors will provide long-lasting or curative treatment, other researchers are betting that new gene-editing technologies, such as CRISPR, will offer the best chance for developing a widespread cure for SCD. One strategy being eyed by these “gene editors” is to correct the SCD mutation, replacing it with a normal gene. Another strategy involves knocking out certain DNA sequences to reactivate production of fetal hemoglobin (HbF).
The HbF protein is produced in the developing fetus to give it better access to oxygen from the mother’s bloodstream. But shortly after birth, the production of fetal hemoglobin shuts down, and the adult form kicks in. Adults normally have very low levels of fetal hemoglobin, which makes sense. However, from genome-wide association studies of human genetic variation, we know that that actual levels of HbF are under genetic control.
A major factor has been mapped to the BCL11A gene, which has subsequently been found to be a master mediator for the fetal to adult hemoglobin switch. Specifically, variations in a red cell specific enhancer of BCL11A affect an adult’s level of HbF— levels of BCL11A protein lead to higher amounts of fetal hemoglobin. Furthermore, it’s been known for some time that rare individuals keep on producing relatively high levels of hemoglobin into adulthood. If people with SCD happen to have a rare mutation that keeps fetal hemoglobin production active in adulthood (the first of these was found as part of my postdoctoral research), their SCD symptoms are much less severe.
Currently, two groups—CRISPR Therapeutics/Vertex Pharmaceuticals and Sangamo Therapeutics/Bioverativ—are gearing up to begin the first U.S. human clinical trials of gene-editing for SCD within the next few months. While they employ different technologies, both approaches involve removing a patient’s HSCs, using gene editing to knock out the BCL11A red cell enhancer, and then returning the gene-edited cells to the patient. The hope is that the gene-edited cells will greatly boost fetal hemoglobin production, thereby offsetting the effects of SCD.
All of this is exciting news for the 100,000 people living in the United States who have SCD. But what about the 300,000 babies born with SCD every year in other parts of the world, mostly in low- and middle-income countries?
The complicated, high-tech procedures that I just described may not be practical for a very long time in places like sub-Saharan Africa. That’s one reason why NIH recently launched a new effort to speed the development of safe, effective genome-editing approaches that could be delivered directly into a patient’s body (in vivo), perhaps by infusion of the CRISPR gene editing apparatus. Recent preclinical experiments demonstrating the promise of in vivo gene editing for Duchenne muscular dystrophy make me optimistic that NIH’s Somatic Cell Genome Editing Program, which is hosting its first gathering of investigators this week, will be able to develop similar approaches for SCD and many other conditions.
While moving forward in this fast-paced field, it is important that we remain ethical, but also remain bold on behalf of the millions of patients with genetic diseases who are still waiting for a cure. We must continue to assess and address the very serious ethical concerns raised by germline gene editing of human embryos, which will irreversibly alter the DNA instruction book of future children and affect future generations. I continue to argue that we are not ready to undertake such experiments.
But the use of gene editing to treat, perhaps even to cure, children and adults with genetic diseases, by correcting the mutation in their relevant tissues (so-called somatic cell gene editing), without risk of passing those changes on to a future generation, holds enormous promise. Somatic cell gene editing is associated with ethical issues that are much more in line with decades of deep thinking about benefits and risks of therapeutic trials.
Finally, we must recognize that somatic cell gene editing is a profoundly promising approach not only for people with SCD, but for all who are struggling with the thousands of diseases that still have no treatments or cures. Real hope for cures has never been greater.
References:
[1] NIH researcher presents encouraging results for gene therapy for severe sickle cell disease. NIH News Release. December 4, 2018
[2] Bluebird bio presents new data for LentiGlobin gene therapy in sickle cell disease at 60th annual meeting of the American Society of Hematology. Bluebird bio. December 3, 2018
Links:
Sickle Cell Disease (National Heart, Lung, and Blood Institute/NIH)
Cure Sickle Cell Initiative (NHLBI)
John Tisdale (NHLBI)
Somatic Cell Genome Editing Program (Common Fund/NIH)
What are genome editing and CRISPR-Cas9? (National Library of Medicine/NIH)
ClinicalTrials.gov (NIH)
NIH Support: National Heart, Lung, and Blood Institute; Common Fund
Sharing a Story of Hope
Posted on by Dr. Francis Collins
Whether by snail mail, email, or social media, it’s the time of year for catching up with family and friends. As NIH Director, I’m also fortunate to hear from some of the amazing people who’ve been helped by NIH research. Among the greetings to arrive in my inbox this holiday season is this incredible video from a 15-year-old named Aaron, who is fortunate enough to count two states—Alabama and Colorado—as his home.
As a young boy, Aaron was naturally athletic, speeding around the baseball diamond and competing on the ski slopes in freestyle mogul. But around the age of 10, Aaron noticed something strange. He couldn’t move as fast as usual. Aaron pushed himself to get back up to speed, but his muscles grew progressively weaker.
Creative Minds: Designing Personalized Clinical Trials
Posted on by Dr. Francis Collins

Karina Davidson/Credit: Jörg Meyer
It might have been 25 years ago, but Karina Davidson remembers that day like yesterday. She was an intern in clinical psychology, and two concerned parents walked into the hospital with their troubled, seven-year-old son. The boy was severely underweight at just 37 pounds and had been acting out violently toward himself and others. It seemed as though Ritalin, a drug commonly prescribed for Attention Deficit Disorder, might help. But would it?
To find out, the clinical team did something unconventional: they designed for the boy a clinical trial to test the benefit of Ritalin versus a placebo. The boy was randomly assigned to take either the drug or placebo each day for four weeks. As a controlled study, neither clinical staff nor the family knew whether he was taking the drug or placebo at any given time. The result: Ritalin wasn’t the answer. The boy was spared any side effects from long term administration of a medication that wouldn’t help him, and his doctors could turn to other potentially more beneficial approaches to his treatment.
Davidson, now an established clinical psychologist at the Columbia University Irving Medical Center, New York, wants to take the unconventional approach that helped this boy and make it more of the norm in medicine. With support from a 2017 NIH Director’s Transformative Research Award, she and her colleagues will develop three pilot computer applications—or digital platforms—to help doctors conduct one-person studies in their offices.
Previous Page Next Page