machine learning
Welcoming Internet Pioneer Vint Cerf for Rall Cultural Lecture on AI in Biomedical Research
Posted on by Dr. Monica M. Bertagnolli

Last week it was my pleasure to welcome to NIH Vinton “Vint” Cerf, a pioneer of the digital world widely known as one of the “fathers of the internet,” to speak at our annual J. Edward Rall Cultural Lecture. We had a lively fireside chat focusing on “The Promises and Perils of AI in Biomedical Research and Health Care Delivery” that included discussions on topics like improving data collection, ensuring broad representativeness of the data being used to train AI and machine learning, expanding clinical research, and offering advice for scientists just starting out. It was an insightful, eye-opening conversation on a topic close to my heart. As NIH Director, I very much hope to deliver evidence-based health care to all people, and I’m excited about how AI and machine learning can help us advance toward that goal.
Dr. Cerf, who is vice president and Chief Internet Evangelist for Google, has too many awards to list, but just a few include the U.S. National Medal of Technology, the Turing Award, and the Presidential Medal of Freedom. He serves as an advisor to several government agencies, including the National Science Foundation, NASA, and the Departments of Defense, Energy, and Commerce.

The NIH Rall Cultural Lecture is held in honor of Dr. J. Edward Rall, who helped define the modern intramural research program at NIH. In the 1950s, he helped establish a stable, academic-minded, and culturally rich community within our rapidly expanding government agency. In 1984, he recommended that NIH add a cultural lecture to its Director’s Lecture series to enrich our scientific community.
As I said at the event, I’m grateful to Dr. Cerf for his visit and this conversation, and I can’t think of a more spectacular Rall Cultural Lecture than what he provided for all of us. You can watch the full talk here.

NIH HEAL Initiative Meets People Where They Are
Posted on by Rebecca Baker, Ph.D., NIH Helping to End Addiction Long-term® (HEAL) Initiative

The opioid crisis continues to devastate communities across America. Dangerous synthetic opioids, like fentanyl, have flooded the illicit drug supply with terrible consequences. Tragically, based on our most-recent data, about 108,000 people in the U.S. die per year from overdoses of opioids or stimulants [1]. Although this complex public health challenge started from our inability to treat pain effectively, chronic pain remains a life-altering problem for 50 million Americans.
To match the size and complexity of the crisis, in 2018 NIH developed the NIH Helping to End Addiction Long-term® (HEAL) Initiative, an aggressive effort involving nearly all of its 27 institutes and centers. Through more than 1,000 research projects, including basic science, clinical testing of new and repurposed drugs, research with communities, and health equity research, HEAL is dedicated to building a new future built on hope.
In this future:
- A predictive tool used during a health visit personalizes treatment for back pain. The tool estimates the probability that a person will benefit from physical therapy, psychotherapy, or surgery.
- Visits to community health clinics and emergency departments serve as routine opportunities to prevent and treat opioid addiction.
- Qualified school staff and pediatricians screen all children for behavioral and other mental health conditions that increase risk for harmful developmental outcomes, including opioid misuse.
- Infants born exposed to opioids during a mother’s pregnancy receive high-quality care—setting them up for a healthy future.
Five years after getting started (and interrupted by a global pandemic), HEAL research is making progress toward achieving this vision. I’ll highlight three ways in which scientific solutions are meeting people where they are today.
A Window of Opportunity for Treatment in the Justice System
Sadly, jails and prisons are “ground zero” for the nation’s opioid crisis. Eighty-five percent of people who are incarcerated have a substance use disorder or a history of substance use. Our vision at HEAL is that every person in jail, prison, or a court-supervised program receives medical care, which includes effective opioid use disorder treatment.
Some research results already are in supporting this approach: A recent HEAL study learned that individuals who had received addiction treatment while in one Massachusetts jail were about 30 percent less likely to be arrested, arraigned, or incarcerated again compared with those incarcerated during the same time period in a neighboring jail that did not offer treatment [2]. Research from the HEAL-supported Justice Community Opioid Innovation Network also is exploring public perceptions about opioid addiction. One such survey showed that most U.S. adults see opioid use disorder as a treatable medical condition rather than as a criminal matter [3]. That’s hopeful news for the future.
A Personalized Treatment Plan for Chronic Back Pain
Half of American adults live with chronic back pain, a major contributor to opioid use. The HEAL-supported Back Pain Consortium (BACPAC) is creating a whole-system model for comprehensive testing of everything that contributes to chronic low back pain, from anxiety to tissue damage. It also includes comprehensive testing of promising pain-management approaches, including psychotherapy, antidepressants, or surgery.
Refining this whole-system model, which is nearing completion, includes finding computer-friendly ways to describe the relationship between the different elements of pain and treatment. That might include developing mathematical equations that describe the physical movements and connections of the vertebrae, discs, and tendons.
Or it might include an artificial intelligence technique called machine learning, in which a computer looks for patterns in existing data, such as electronic health records or medical images. In keeping with HEAL’s all-hands-on-deck approach, BACPAC also conducts clinical trials to test new (or repurposed) treatments and develop new technologies focused on back pain, like a “wearable muscle” to help support the back.
Harnessing Innovation from the Private Sector
The HEAL research portfolio spans basic science to health services research. That allows us to put many shots on goal that will need to be commercialized to help people. Through its research support of small businesses, HEAL funding offers a make-or-break opportunity to advance a great idea to the marketplace, providing a bridge to venture capital or other larger funding sources needed for commercialization.
This bridge also allows HEAL to invest directly in the heart of innovation. Currently, HEAL funds nearly 100 such companies across 20 states. While this is a relatively small portion of all HEAL research, it is science that will make a difference in our communities, and these researchers are passionate about what they do to build a better future.
A couple of current examples of this research passion include: delivery of controlled amounts of non-opioid pain medications after surgery using a naturally absorbable film or a bone glue; immersive virtual reality to help people with opioid use disorder visualize the consequences of certain personal choices; and mobile apps that support recovery, taking medications, or sensing an overdose.
In 2023, HEAL is making headway toward its mission to accelerate development of safe, non-addictive, and effective strategies to prevent and treat pain, opioid misuse, and overdose. We have 314 clinical trials underway and 41 submissions to the Food and Drug Administration to begin clinical testing of investigational new drugs or devices: That number has doubled in the last year. More than 100 projects alone are addressing back pain, and more than 200 projects are studying medications for opioid use disorder.
The nation’s opioid crisis is profoundly difficult and multifaceted—and it won’t be solved with any single approach. Our research is laser-focused on its vision of ending addiction long-term, including improving pain management and expanding access to underused, but highly effective, addiction medications. Every day, we imagine a better future for people with physical and emotional pain and communities that are hurting. Hundreds of researchers and community members across the country are working to achieve a future where people and communities have the tools they need to thrive.
References:
[1] Provisional drug overdose death counts. Ahmad FB, Cisewski JA, Rossen LM, Sutton P. National Center for Health Statistics. 2023.
[2] Recidivism and mortality after in-jail buprenorphine treatment for opioid use disorder. Evans EA, Wilson D, Friedmann PD. Drug Alcohol Depend. 2022 Feb 1;231:109254.
[3] Social stigma toward persons with opioid use disorder: Results from a nationally representative survey of U.S. adults. Taylor BG, Lamuda PA, Flanagan E, Watts E, Pollack H, Schneider J. Subst Use Misuse. 2021;56(12):1752-1764.
Links:
SAMHSA’s National Helpline (Substance Abuse and Mental Health Services Administration, Rockville, MD)
NIH Helping to End Addiction Long-term® (HEAL) Initiative
Video: The NIH HEAL Initiative–HEAL Is Hope
Justice Community Opioid Innovation Network (HEAL)
Back Pain Consortium Research Program (HEAL)
NIH HEAL Initiative 2023 Annual Report (HEAL)
Small Business Programs (HEAL)
Rebecca Baker (HEAL)
Note: Dr. Lawrence Tabak, who performs the duties of the NIH Director, has asked the heads of NIH’s Institutes, Centers, and Offices to contribute occasional guest posts to the blog to highlight some of the interesting science that they support and conduct. This is the 28th in the series of NIH guest posts that will run until a new permanent NIH director is in place.
A Look Back at Science’s 2022 Breakthroughs
Posted on by Lawrence Tabak, D.D.S., Ph.D.
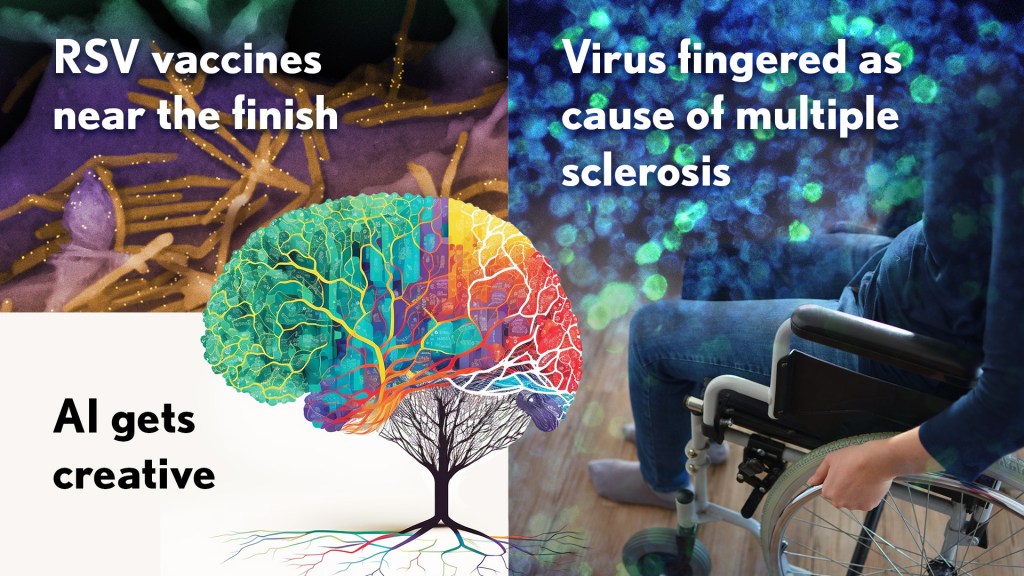
Happy New Year! I hope everyone finished 2022 with plenty to celebrate, whether it was completing a degree or certification, earning a promotion, attaining a physical fitness goal, or publishing a hard-fought scientific discovery.
If the latter, you are in good company. Last year produced some dazzling discoveries, and the news and editorial staff at the journal Science kept a watchful eye on the most high-impact advances of 2022. In December, the journal released its list of the top 10 advances across the sciences, from astronomy to zoology. In case you missed it, Science selected NASA’s James Webb Space Telescope (JWST) as the 2022 Breakthrough of the Year [1].
This unique space telescope took 20 years to complete, but it has turned out to be time well spent. Positioned 1.5-million-kilometers from Earth, the JWST and its unprecedented high-resolution images of space have unveiled the universe anew for astronomers and wowed millions across the globe checking in online. The telescope’s image stream, beyond its sheer beauty, will advance study of the early Universe, allowing astronomers to discover distant galaxies, explore the early formation of stars, and investigate the possibility of life on other planets.
While the biomedical sciences didn’t take home the top prize, they were well represented among Science’s runner-up breakthroughs. Some of these biomedical top contenders also have benefited, directly or indirectly, from NIH efforts and support. Let’s take a look:
RSV vaccines nearing the finish line: It’s been one of those challenging research marathons. But scientists last year started down the homestretch with the first safe-and-effective vaccine for respiratory syncytial virus (RSV), a leading cause of severe respiratory illness in the very young and the old.
In August, the company Pfizer presented evidence that its experimental RSV vaccine candidate offered protection for those age 60 and up. Later, they showed that the same vaccine, when administered to pregnant women, helped to protect their infants against RSV for six months after birth. Meanwhile, in October, the company GSK announced encouraging results from its late-stage phase III trial of an RSV vaccine in older adults.
As Science noted, the latest clinical progress also shows the power of basic science. For example, researchers have been working with chemically inactivated versions of the virus to develop the vaccine. But these versions have a key viral surface protein that changes its shape after fusing with a cell to start an infection. In this configuration, the protein elicits only weak levels of needed protective antibodies.
Back in 2013, Barney Graham, then with NIH’s National Institute of Allergy and Infectious Diseases (NIAID), and colleagues, solved the problem [2]. Graham’s NIH team discovered a way to lock the protein into its original prefusion state, which the immune system can better detect. This triggers higher levels of potent antibodies, and the discovery kept the science—and the marathon—moving forward.
These latest clinical advances come as RSV and other respiratory viruses, including SARS-CoV-2, the cause of COVID-19, are sending an alarming number of young children to the hospital. The hope is that researchers will cross the finish line this year or next, and we’ll have the first approved RSV vaccine.
Virus fingered as cause of multiple sclerosis: Researchers have long thought that multiple sclerosis, or MS, has a viral cause. Pointing to the right virus with the required high degree of certainty has been the challenge, slowing progress on the treatment front for those in need. As published in Science last January, Alberto Ascherio, Harvard T.H. Chan School of Public Health, Boston, and colleagues produced the strongest evidence yet that MS is caused by the Epstein-Barr virus (EBV), a herpesvirus also known for causing infectious mononucleosis [3].
The link between EBV and MS had long been suspected. But it was difficult to confirm because EBV infections are so widespread, and MS is so disproportionately rare. In the recent study, the NIH-supported researchers collected blood samples every other year from more than 10 million young adults in the U.S. military, including nearly 1,000 who were diagnosed with MS during their service. The evidence showed that the risk of an MS diagnosis increased 32-fold after EBV infection, but it held steady following infection with any other virus. Levels in blood serum of a biomarker for MS neurodegeneration also went up only after an EBV infection, suggesting that the viral illness is a leading cause for MS.
Further evidence came last year from a discovery published in the journal Nature by William Robinson, Stanford University School of Medicine, Stanford, CA, and colleagues. The NIH-supported team found a close resemblance between an EBV protein and one made in the healthy brain and spinal cord [4]. The findings suggest an EBV infection may produce antibodies that mistakenly attack the protective sheath surrounding our nerve cells. Indeed, the study showed that up to one in four people with MS had antibodies that bind both proteins.
This groundbreaking research suggests that an EBV vaccine and/or antiviral drugs that thwart this infection might ultimately prevent or perhaps even cure MS. Of note, NIAID launched last May an early-stage clinical trial for an experimental EBV vaccine at the NIH Clinical Center, Bethesda, MD.
AI Gets Creative: Science’s 2021 Breakthrough of the Year was AI-powered predictions of protein structure. In 2022, AI returned to take another well-deserved bow. This time, Science singled out AI’s now rapidly accelerating entry into once uniquely human attributes, such as artistic expression and scientific discovery.
On the scientific discovery side, Science singled out AI’s continued progress in getting creative with the design of novel proteins for vaccines and myriad other uses. One technique, called “hallucination,” generates new proteins from scratch. Researchers input random amino acid sequences into the computer, and it randomly and continuously mutates them into sequences that other AI tools are confident will fold into stable proteins. This greatly simplifies the process of protein design and frees researchers to focus their efforts on creating a protein with a desired function.
AI research now engages scientists around world, including hundreds of NIH grantees. Taking a broader view of AI, NIH recently launched the Artificial Intelligence/Machine Learning Consortium to Advance Health Equity and Researcher Diversity (AIM-AHEAD) Program. It will help to create greater diversity within the field, which is a must. A lack of diversity could perpetuate harmful biases in how AI is used, how algorithms are developed and trained, and how findings are interpreted to avoid health disparities and inequities for underrepresented communities.
And there you have it, some of the 2022 breakthroughs from Science‘s news and editorial staff. Of course, the highlighted biomedical breakthroughs don’t capture the full picture of research progress. There were many other milestone papers published in 2022 that researchers worldwide will build upon in the months and years ahead to make further progress in their disciplines and, for some, draw the attention of Science’s news and editorial staff. Here’s to another productive year in biomedical research, which the blog will continue to feature and share with you as it unfolds in 2023.
References:
[1] 2022 Breakthrough of the Year. Science. Dec 15, 2022.
[2] Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. McLellan JS, Chen M, Leung S, Kwong PD, Graham BS, et al. Science. 2013 May 31;340(6136):1113-1117.
[3] Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, Elledge SJ, Niebuhr DW, Scher AI, Munger KL, Ascherio A. Science. 2022 Jan 21;375(6578):296-301.
[4] Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Lanz TV, Brewer RC, Steinman L, Robinson WH, et al. Nature. 2022 Mar;603(7900):321-327.
Links:
Respiratory Syncytial Virus (RSV) (National Institute of Allergy and Infectious Diseases/NIH)
Multiple Sclerosis (National Institute of Neurological Disorders and Stroke/NIH)
Barney Graham (Morehouse School of Medicine, Atlanta)
Alberto Ascherio (Harvard T.H. Chan School of Public Health, Boston)
Robinson Lab (Stanford Medicine, Stanford, CA)
James Webb Space Telescope (Goddard Space Flight Center/NASA, Greenbelt, MD)
National Library of Medicine Helps Lead the Way in AI Research
Posted on by Patricia Flatley Brennan, R.N., Ph.D., National Library of Medicine
Did you know that the NIH’s National Library of Medicine (NLM) has been serving science and society since 1836? From its humble beginning as a small collection of books in the library of the U.S. Army Surgeon General’s office, NLM has grown not only to become the world’s largest biomedical library, but a leader in biomedical informatics and computational health data science research.
Think of NLM as a door through which you pass to connect with health data, literature, medical and scientific information, expertise, and sophisticated mathematical models or images that describe a clinical problem. This intersection of information, people, and technology allows NLM to foster discovery. NLM does so by ensuring that scientists, clinicians, librarians, patients, and the public have access to biomedical information 24 hours a day, 7 days a week.
The NLM also supports two research efforts: the Division of Extramural Programs (EP) and Intramural Research Program (IRP). Both programs are accelerating advances in biomedical informatics, data science, computational biology, and computational health. One of EP’s notable investments is focused on advancing artificial intelligence (AI) methods and reimagining how health care is delivered with the power of AI.

With support from NLM, Corey Lester and his colleagues at the University of Michigan College of Pharmacy, Ann Arbor, MI, are using AI to assist in pill verification, a standard procedure in pharmacies across the land. They want to help pharmacists avoid dangerous and costly dispensing errors. To do so, Lester is using AI to develop a real-time computer vision model. It views pills inside of a medication bottle, accurately identifies them, and determines that they are the correct or incorrect contents.
The IRP develops and applies computational methods and approaches to a broad range of information problems in biology, biomedicine, and human health. The IRP also offers intramural training opportunities and supports other training aimed at pre-baccalaureate to postdoctoral students and professionals.
The NLM principal investigators use biological data to advance computer algorithms and connect relationships between any level of biological organization and health conditions. They also use computational health sciences to focus on clinical information processing and analyze clinical data, assess clinical outcomes, and set health data standards.

NLM investigator Sameer Antani is collaborating with researchers in other NIH institutes to explore how AI can help us understand oral cancer, echocardiography, and pediatric tuberculosis. His research also is examining how images can be mined for data to predict the causes and outcomes of conditions. Examples of Antani’s work can be found in mobile radiology vehicles, which allow professionals to take chest X-rays (right) and screen for HIV and tuberculosis using software containing algorithms developed in his lab.
For AI to have its full impact, more algorithms and approaches that harness the power of data are needed. That’s why NLM supports hundreds of other intramural and extramural scientists who are addressing challenging health and biomedical problems. The NLM-funded research is focused on how AI can help people stay healthy through early disease detection, disease management, and clinical and treatment decision-making—all leading to the ultimate goal of helping people live healthier and happier lives.
The NLM is proud to lead the way in the use of AI to accelerate discovery and transform health care. Want to learn more? Follow me on Twitter. Or, you can follow my blog, NLM Musings from the Mezzanine and receive periodic NLM research updates.
I would like to thank Valerie Florance, Acting Scientific Director of NLM IRP, and Richard Palmer, Acting Director of NLM Division of EP, for their assistance with this post.
Links:
National Library of Medicine (National Library of Medicine/NIH)
Video: Using Machine Intelligence to Prevent Medication Dispensing Errors (NLM Funding Spotlight)
Video: Sameer Antani and Artificial Intelligence (NLM)
NLM Division of Extramural Programs (NLM)
NLM Intramural Research Program (NLM)
NLM Intramural Training Opportunities (NLM)
Principal Investigators (NLM)
NLM Musings from the Mezzanine (NLM)
Note: Dr. Lawrence Tabak, who performs the duties of the NIH Director, has asked the heads of NIH’s Institutes and Centers (ICs) to contribute occasional guest posts to the blog to highlight some of the interesting science that they support and conduct. This is the 20th in the series of NIH IC guest posts that will run until a new permanent NIH director is in place.
Understanding Long-Term COVID-19 Symptoms and Enhancing Recovery
Posted on by Walter J. Koroshetz, M.D., National Institute of Neurological Disorders and Stroke

We are in the third year of the COVID-19 pandemic, and across the world, most restrictions have lifted, and society is trying to get back to “normal.” But for many people—potentially millions globally—there is no getting back to normal just yet.
They are still living with the long-term effects of a COVID-19 infection, known as the post-acute sequelae of SARS-CoV-2 infection (PASC), including Long COVID. These people continue to experience debilitating fatigue, shortness of breath, pain, difficulty sleeping, racing heart rate, exercise intolerance, gastrointestinal and other symptoms, as well as cognitive problems that make it difficult to perform at work or school.
This is a public health issue that is in desperate need of answers. Research is essential to address the many puzzling aspects of Long COVID and guide us to effective responses that protect the nation’s long-term health.
For the past two years, NIH’s National Heart, Lung, and Blood Institute (NHLBI), the National Institute of Allergy and Infectious Diseases (NIAID), and my National Institute of Neurological Disorders and Stroke (NINDS) along with several other NIH institutes and the office of the NIH Director, have been leading NIH’s Researching COVID to Enhance Recovery (RECOVER) initiative, a national research program to understand PASC.
The initiative studies core questions such as why COVID-19 infections can have lingering effects, why new symptoms may develop, and what is the impact of SARS-CoV-2, the virus that causes COVID-19, on other diseases and conditions? Answering these fundamental questions will help to determine the underlying biologic basis of Long COVID. The answers will also help to tell us who is at risk for Long COVID and identify therapies to prevent or treat the condition.
The RECOVER initiative’s wide scope of research is also unprecedented. It is needed because Long COVID is so complex, and history indicates that similar post infectious conditions have defied definitive explanation or effective treatment. Indeed, those experiencing Long COVID report varying symptoms, making it highly unlikely that a single therapy will work for everyone, underscoring the need to pursue multiple therapeutic strategies.
To understand Long COVID fully, hundreds of RECOVER investigators are recruiting more than 17,000 adults (including pregnant people) and more than 18,000 children to take part in cohort studies. Hundreds of enrolling sites have been set up across the country. An autopsy research cohort will also provide further insight into how COVID-19 affects the body’s organs and tissues.
In addition, researchers will analyze electronic health records from millions of people to understand how Long COVID and its symptoms change over time. The RECOVER initiative is also utilizing consistent research protocols across all the study sites. The protocols have been carefully developed with input from patients and advocates, and they are designed to allow for consistent data collection, improve data sharing, and help to accelerate the pace of research.
From the very beginning, people suffering from Long COVID have been our partners in RECOVER. Patients and advocates have contributed important perspectives and provided valuable input into the master protocols and research plans.
Now, with RECOVER underway, individuals with Long COVID, their caregivers, and community members continue to serve a critical role in the Initiative. The National Community Engagement Group (NCEG) has been established to make certain that RECOVER meets the needs of all people affected by Long COVID. The RECOVER Patient and Community Engagement Strategy outlines all the approaches that RECOVER is using to engage with and gather input from individuals impacted by Long COVID.
The NIH recently made more than 40 awards to improve understanding of the underlying biology and pathology of Long COVID. There have already been several important findings published by RECOVER scientists.
For example, in a recent study published in the journal Lancet Digital Health, RECOVER investigators used machine learning to comb through electronic health records to look for signals that may predict whether someone has Long COVID [1]. As new findings, tools, and technologies continue to emerge that help advance our knowledge of the condition, the RECOVER Research Review (R3) Seminar Series will provide a forum for researchers and our partners with up-to-date information about Long COVID research.
It is important to note that post-viral conditions are not a new concept. Many, but not all, of the symptoms reported in Long COVID, including fatigue, post-exertional malaise, chronic musculoskeletal pain, sleep disorders, postural orthostatic tachycardia (POTS), and cognitive issues, overlap with myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS).
ME/CFS is a serious disease that can occur following infection and make people profoundly sick for decades. Like Long COVID, ME/CFS is a heterogenous condition that does not affect everybody in the same way, and the knowledge gained through research on Long COVID may also positively impact the understanding, treatment, and prevention of POTS, ME/CFS, and other chronic diseases.
Unlike other post-viral conditions, people who experience Long COVID were all infected by the same virus—albeit different variants—at a similar point in time. This creates a unique opportunity for RECOVER researchers to study post-viral conditions in real-time.
The opportunity enables scientists to study many people simultaneously while they are still infected to monitor their progress and recovery, and to try to understand why some individuals develop ongoing symptoms. A better understanding of the transition from acute to chronic disease may offer an opportunity to intervene, identify who is at risk of the transition, and develop therapies for people who experience symptoms long after the acute infection has resolved.
The RECOVER initiative will soon announce clinical trials, leveraging data from clinicians and patients in which symptom clusters were identified and can be targeted by various interventions. These trials will investigate therapies that are indicated for other non-COVID conditions and novel treatments for Long COVID.
Through extensive collaboration across the multiple NIH institutes and offices that contribute to the RECOVER effort, our hope is critical answers will emerge soon. These answers will help us to recognize the full range of outcomes and needs resulting from PASC and, most important, enable many people to make a full recovery from COVID-19. We are indebted to the over 10,000 subjects who have already enrolled in RECOVER. Their contributions and the hard work of the RECOVER investigators offer hope for the future to the millions still suffering from the pandemic.
Reference:
[1] Identifying who has long COVID in the USA: a machine learning approach using N3C data. Pfaff ER, Girvin AT, Bennett TD, Bhatia A, Brooks IM, Deer RR, Dekermanjian JP, Jolley SE, Kahn MG, Kostka K, McMurry JA, Moffitt R, Walden A, Chute CG, Haendel MA; N3C Consortium. Lancet Digit Health. 2022 Jul;4(7):e532-e541.
Links:
COVID-19 Research (NIH)
Long COVID (NIH)
RECOVER: Researching COVID to Enhance Recovery (NIH)
“NIH builds large nationwide study population of tens of thousands to support research on long-term effects of COVID-19,” NIH News Release, September 15, 2021.
Director’s Messages (National Institute of Neurological Disorders and Stroke/NIH)
Note: Dr. Lawrence Tabak, who performs the duties of the NIH Director, has asked the heads of NIH’s Institutes and Centers (ICs) to contribute occasional guest posts to the blog to highlight some of the interesting science that they support and conduct. This is the 18th in the series of NIH IC guest posts that will run until a new permanent NIH director is in place.
Using AI to Find New Antibiotics Still a Work in Progress
Posted on by Lawrence Tabak, D.D.S., Ph.D.
Each year, more than 2.8 million people in the United States develop bacterial infections that don’t respond to treatment and sometimes turn life-threatening [1]. Their infections are antibiotic-resistant, meaning the bacteria have changed in ways that allow them to withstand our current widely used arsenal of antibiotics. It’s a serious and growing health-care problem here and around the world. To fight back, doctors desperately need new antibiotics, including novel classes of drugs that bacteria haven’t seen and developed ways to resist.
Developing new antibiotics, however, involves much time, research, and expense. It’s also fraught with false leads. That’s why some researchers have turned to harnessing the predictive power of artificial intelligence (AI) in hopes of selecting the most promising leads faster and with greater precision.
It’s a potentially paradigm-shifting development in drug discovery, and a recent NIH-funded study, published in the journal Molecular Systems Biology, demonstrates AI’s potential to streamline the process of selecting future antibiotics [2]. The results are also a bit sobering. They highlight the current limitations of one promising AI approach, showing that further refinement will still be needed to maximize its predictive capabilities.
These findings come from the lab of James Collins, Massachusetts Institute of Technology (MIT), Cambridge, and his recently launched Antibiotics-AI Project. His audacious goal is to develop seven new classes of antibiotics to treat seven of the world’s deadliest bacterial pathogens in just seven years. What makes this project so bold is that only two new classes of antibiotics have reached the market in the last 50 years!
In the latest study, Collins and his team looked to an AI program called AlphaFold2 [3]. The name might ring a bell. AlphaFold’s AI-powered ability to predict protein structures was a finalist in Science Magazine’s 2020 Breakthrough of the Year. In fact, AlphaFold has been used already to predict the structures of more than 200 million proteins, or almost every known protein on the planet [4].
AlphaFold employs a deep learning approach that can predict most protein structures from their amino acid sequences about as well as more costly and time-consuming protein-mapping techniques.
In the deep learning models used to predict protein structure, computers are “trained” on existing data. As computers “learn” to understand complex relationships within the training material, they develop a model that can then be applied for making predictions of 3D protein structures from linear amino acid sequences without relying on new experiments in the lab.
Collins and his team hoped to combine AlphaFold with computer simulations commonly used in drug discovery as a way to predict interactions between essential bacterial proteins and antibacterial compounds. If it worked, researchers could then conduct virtual rapid screens of millions of new synthetic drug compounds targeting key bacterial proteins that existing antibiotics don’t. It would also enable the rapid development of antibiotics that work in novel ways, exactly what doctors need to treat antibiotic-resistant infections.
To test the strategy, Collins and his team focused first on the predicted structures of 296 essential proteins from the Escherichia coli bacterium as well as 218 antibacterial compounds. Their computer simulations then predicted how strongly any two molecules (essential protein and antibacterial) would bind together based on their shapes and physical properties.
It turned out that screening many antibacterial compounds against many potential targets in E. coli led to inaccurate predictions. For example, when comparing their computational predictions with actual interactions for 12 essential proteins measured in the lab, they found that their simulated model had about a 50:50 chance of being right. In other words, it couldn’t identify true interactions between drugs and proteins any better than random guessing.
They suspect one reason for their model’s poor performance is that the protein structures used to train the computer are fixed, not flexible and shifting physical configurations as happens in real life. To improve their success rate, they ran their predictions through additional machine-learning models that had been trained on data to help them “learn” how proteins and other molecules reconfigure themselves and interact. While this souped-up model got somewhat better results, the researchers report that they still aren’t good enough to identify promising new drugs and their protein targets.
What now? In future studies, the Collins lab will continue to incorporate and train the computers on even more biochemical and biophysical data to help with the predictive process. That’s why this study should be interpreted as an interim progress report on an area of science that will only get better with time.
But it’s also a sobering reminder that the quest to find new classes of antibiotics won’t be easy—even when aided by powerful AI approaches. We certainly aren’t there yet, but I’m confident that we will get there to give doctors new therapeutic weapons and turn back the rise in antibiotic-resistant infections.
References:
[1] 2019 Antibiotic resistance threats report. Centers for Disease Control and Prevention.
[2] Benchmarking AlphaFold-enabled molecular docking predictions for antibiotic discovery. Wong F, Krishnan A, Zheng EJ, Stark H, Manson AL, Earl AM, Jaakkola T, Collins JJ. Molecular Systems Biology. 2022 Sept 6. 18: e11081.
[3] Highly accurate protein structure prediction with AlphaFold. Jumper J, Evans R, Pritzel A, Kavukcuoglu K, Kohli P, Hassabis D., et al. Nature. 2021 Aug;596(7873):583-589.
[4] ‘The entire protein universe’: AI predicts shape of nearly every known protein. Callaway E. Nature. 2022 Aug;608(7921):15-16.
Links:
Antimicrobial (Drug) Resistance (National Institute of Allergy and Infectious Diseases/NIH)
Collins Lab (Massachusetts Institute of Technology, Cambridge)
The Antibiotics-AI Project, The Audacious Project (TED)
AlphaFold (Deep Mind, London, United Kingdom)
NIH Support: National Institute of Allergy and Infectious Diseases; National Institute of General Medical Sciences
Using AI to Advance Understanding of Long COVID Syndrome
Posted on by Lawrence Tabak, D.D.S., Ph.D.

The COVID-19 pandemic continues to present considerable public health challenges in the United States and around the globe. One of the most puzzling is why many people who get over an initial and often relatively mild COVID illness later develop new and potentially debilitating symptoms. These symptoms run the gamut including fatigue, shortness of breath, brain fog, anxiety, and gastrointestinal trouble.
People understandably want answers to help them manage this complex condition referred to as Long COVID syndrome. But because Long COVID is so variable from person to person, it’s extremely difficult to work backwards and determine what these people had in common that might have made them susceptible to Long COVID. The variability also makes it difficult to identify all those who have Long COVID, whether they realize it or not. But a recent study, published in the journal Lancet Digital Health, shows that a well-trained computer and its artificial intelligence can help.
Researchers found that computers, after scanning thousands of electronic health records (EHRs) from people with Long COVID, could reliably make the call. The results, though still preliminary and in need of further validation, point the way to developing a fast, easy-to-use computer algorithm to help determine whether a person with a positive COVID test is likely to battle Long COVID.
In this groundbreaking study, NIH-supported researchers led by Emily Pfaff, University of North Carolina, Chapel Hill, and Melissa Haendel, the University of Colorado Anschutz Medical Campus, Aurora, relied on machine learning. In machine learning, a computer sifts through vast amounts of data to look for patterns. One reason machine learning is so powerful is that it doesn’t require humans to tell the computer which features it should look for. As such, machine learning can pick up on subtle patterns that people would otherwise miss.
In this case, Pfaff, Haendel, and team decided to “train” their computer on EHRs from people who had reported a COVID-19 infection. (The records are de-identified to protect patient privacy.) The researchers found just what they needed in the National COVID Cohort Collaborative (N3C), a national, publicly available data resource sponsored by NIH’s National Center for Advancing Translational Sciences. It is part of NIH’s Researching COVID to Enhance Recovery (RECOVER) initiative, which aims to improve understanding of Long COVID.
The researchers defined a group of more than 1.5 million adults in N3C who either had been diagnosed with COVID-19 or had a record of a positive COVID-19 test at least 90 days prior. Next, they examined common features, including any doctor visits, diagnoses, or medications, from the group’s roughly 100,000 adults.
They fed that EHR data into a computer, along with health information from almost 600 patients who’d been seen at a Long COVID clinic. They developed three machine learning models: one to identify potential long COVID patients across the whole dataset and two others that focused separately on people who had or hadn’t been hospitalized.
All three models proved effective for identifying people with potential Long-COVID. Each of the models had an 85 percent or better discrimination threshold, indicating they are highly accurate. That’s important because, once researchers can identify those with Long COVID in a large database of people such as N3C, they can begin to ask and answer many critical questions about any differences in an individual’s risk factors or treatment that might explain why some get Long COVID and others don’t.
This new study is also an excellent example of N3C’s goal to assemble data from EHRs that enable researchers around the world to get rapid answers and seek effective interventions for COVID-19, including its long-term health effects. It’s also made important progress toward the urgent goal of the RECOVER initiative to identify people with or at risk for Long COVID who may be eligible to participate in clinical trials of promising new treatment approaches.
Long COVID remains a puzzling public health challenge. Another recent NIH study published in the journal Annals of Internal Medicine set out to identify people with symptoms of Long COVID, most of whom had recovered from mild-to-moderate COVID-19 [2]. More than half had signs of Long COVID. But, despite extensive testing, the NIH researchers were unable to pinpoint any underlying cause of the Long COVID symptoms in most cases.
So if you’d like to help researchers solve this puzzle, RECOVER is now enrolling adults and kids—including those who have and have not had COVID—at more than 80 study sites around the country.
References:
[1] Identifying who has long COVID in the USA: a machine learning approach using N3C data. Pfaff ER, Girvin AT, Bennett TD, Bhatia A, Brooks IM, Deer RR, Dekermanjian JP, Jolley SE, Kahn MG, Kostka K, McMurry JA, Moffitt R, Walden A, Chute CG, Haendel MA; N3C Consortium. Lancet Digit Health. 2022 May 16:S2589-7500(22)00048-6.
[2] A longitudinal study of COVID-19 sequelae and immunity: baseline findings. Sneller MC, Liang CJ, Marques AR, Chung JY, Shanbhag SM, Fontana JR, Raza H, Okeke O, Dewar RL, Higgins BP, Tolstenko K, Kwan RW, Gittens KR, Seamon CA, McCormack G, Shaw JS, Okpali GM, Law M, Trihemasava K, Kennedy BD, Shi V, Justement JS, Buckner CM, Blazkova J, Moir S, Chun TW, Lane HC. Ann Intern Med. 2022 May 24:M21-4905.
Links:
COVID-19 Research (NIH)
National COVID Cohort Collaborative (N3C) (National Center for Advancing Translational Sciences/NIH)
Emily Pfaff (University of North Carolina, Chapel Hill)
Melissa Haendel (University of Colorado, Aurora)
NIH Support: National Center for Advancing Translational Sciences; National Institute of General Medical Sciences; National Institute of Allergy and Infectious Diseases
Millions of Single-Cell Analyses Yield Most Comprehensive Human Cell Atlas Yet
Posted on by Lawrence Tabak, D.D.S., Ph.D.

There are 37 trillion or so cells in our bodies that work together to give us life. But it may surprise you that we still haven’t put a good number on how many distinct cell types there are within those trillions of cells.
That’s why in 2016, a team of researchers from around the globe launched a historic project called the Human Cell Atlas (HCA) consortium to identify and define the hundreds of presumed distinct cell types in our bodies. Knowing where each cell type resides in the body, and which genes each one turns on or off to create its own unique molecular identity, will revolutionize our studies of human biology and medicine across the board.
Since its launch, the HCA has progressed rapidly. In fact, it has already reached an important milestone with the recent publication in the journal Science of four studies that, together, comprise the first multi-tissue drafts of the human cell atlas. This draft, based on analyses of millions of cells, defines more than 500 different cell types in more than 30 human tissues. A second draft, with even finer definition, is already in the works.
Making the HCA possible are recent technological advances in RNA sequencing. RNA sequencing is a topic that’s been mentioned frequently on this blog in a range of research areas, from neuroscience to skin rashes. Researchers use it to detect and analyze all the messenger RNA (mRNA) molecules in a biological sample, in this case individual human cells from a wide range of tissues, organs, and individuals who voluntarily donated their tissues.
By quantifying these RNA messages, researchers can capture the thousands of genes that any given cell actively expresses at any one time. These precise gene expression profiles can be used to catalogue cells from throughout the body and understand the important similarities and differences among them.
In one of the published studies, funded in part by the NIH, a team co-led by Aviv Regev, a founding co-chair of the consortium at the Broad Institute of MIT and Harvard, Cambridge, MA, established a framework for multi-tissue human cell atlases [1]. (Regev is now on leave from the Broad Institute and MIT and has recently moved to Genentech Research and Early Development, South San Francisco, CA.)
Among its many advances, Regev’s team optimized single-cell RNA sequencing for use on cell nuclei isolated from frozen tissue. This technological advance paved the way for single-cell analyses of the vast numbers of samples that are stored in research collections and freezers all around the world.
Using their new pipeline, Regev and team built an atlas including more than 200,000 single-cell RNA sequence profiles from eight tissue types collected from 16 individuals. These samples were archived earlier by NIH’s Genotype-Tissue Expression (GTEx) project. The team’s data revealed unexpected differences among cell types but surprising similarities, too.
For example, they found that genetic profiles seen in muscle cells were also present in connective tissue cells in the lungs. Using novel machine learning approaches to help make sense of their data, they’ve linked the cells in their atlases with thousands of genetic diseases and traits to identify cell types and genetic profiles that may contribute to a wide range of human conditions.
By cross-referencing 6,000 genes previously implicated in causing specific genetic disorders with their single-cell genetic profiles, they identified new cell types that may play unexpected roles. For instance, they found some non-muscle cells that may play a role in muscular dystrophy, a group of conditions in which muscles progressively weaken. More research will be needed to make sense of these fascinating, but vital, discoveries.
The team also compared genes that are more active in specific cell types to genes with previously identified links to more complex conditions. Again, their data surprised them. They identified new cell types that may play a role in conditions such as heart disease and inflammatory bowel disease.
Two of the other papers, one of which was funded in part by NIH, explored the immune system, especially the similarities and differences among immune cells that reside in specific tissues, such as scavenging macrophages [2,3] This is a critical area of study. Most of our understanding of the immune system comes from immune cells that circulate in the bloodstream, not these resident macrophages and other immune cells.
These immune cell atlases, which are still first drafts, already provide an invaluable resource toward designing new treatments to bolster immune responses, such as vaccines and anti-cancer treatments. They also may have implications for understanding what goes wrong in various autoimmune conditions.
Scientists have been working for more than 150 years to characterize the trillions of cells in our bodies. Thanks to this timely effort and its advances in describing and cataloguing cell types, we now have a much better foundation for understanding these fundamental units of the human body.
But the latest data are just the tip of the iceberg, with vast flows of biological information from throughout the human body surely to be released in the years ahead. And while consortium members continue making history, their hard work to date is freely available to the scientific community to explore critical biological questions with far-reaching implications for human health and disease.
References:
[1] Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Eraslan G, Drokhlyansky E, Anand S, Fiskin E, Subramanian A, Segrè AV, Aguet F, Rozenblatt-Rosen O, Ardlie KG, Regev A, et al. Science. 2022 May 13;376(6594):eabl4290.
[2] Cross-tissue immune cell analysis reveals tissue-specific features in humans. Domínguez Conde C, Xu C, Jarvis LB, Rainbow DB, Farber DL, Saeb-Parsy K, Jones JL,Teichmann SA, et al. Science. 2022 May 13;376(6594):eabl5197.
[3] Mapping the developing human immune system across organs. Suo C, Dann E, Goh I, Jardine L, Marioni JC, Clatworthy MR, Haniffa M, Teichmann SA, et al. Science. 2022 May 12:eabo0510.
Links:
Ribonucleic acid (RNA) (National Human Genome Research Institute/NIH)
Studying Cells (National Institute of General Medical Sciences/NIH)
Regev Lab (Broad Institute of MIT and Harvard, Cambridge, MA)
NIH Support: Common Fund; National Cancer Institute; National Human Genome Research Institute; National Heart, Lung, and Blood Institute; National Institute on Drug Abuse; National Institute of Mental Health; National Institute on Aging; National Institute of Allergy and Infectious Diseases; National Institute of Neurological Disorders and Stroke; National Eye Institute
Artificial Intelligence Accurately Predicts RNA Structures, Too
Posted on by Dr. Francis Collins

Researchers recently showed that a computer could “learn” from many examples of protein folding to predict the 3D structure of proteins with great speed and precision. Now a recent study in the journal Science shows that a computer also can predict the 3D shapes of RNA molecules [1]. This includes the mRNA that codes for proteins and the non-coding RNA that performs a range of cellular functions.
This work marks an important basic science advance. RNA therapeutics—from COVID-19 vaccines to cancer drugs—have already benefited millions of people and will help many more in the future. Now, the ability to predict RNA shapes quickly and accurately on a computer will help to accelerate understanding these critical molecules and expand their healthcare uses.
Like proteins, the shapes of single-stranded RNA molecules are important for their ability to function properly inside cells. Yet far less is known about these RNA structures and the rules that determine their precise shapes. The RNA elements (bases) can form internal hydrogen-bonded pairs, but the number of possible combinations of pairings is almost astronomical for any RNA molecule with more than a few dozen bases.
In hopes of moving the field forward, a team led by Stephan Eismann and Raphael Townshend in the lab of Ron Dror, Stanford University, Palo Alto, CA, looked to a machine learning approach known as deep learning. It is inspired by how our own brain’s neural networks process information, learning to focus on some details but not others.
In deep learning, computers look for patterns in data. As they begin to “see” complex relationships, some connections in the network are strengthened while others are weakened.
One of the things that makes deep learning so powerful is it doesn’t rely on any preconceived notions. It also can pick up on important features and patterns that humans can’t possibly detect. But, as successful as this approach has been in solving many different kinds of problems, it has primarily been applied to areas of biology, such as protein folding, in which lots of data were available for researchers to train the computers.
That’s not the case with RNA molecules. To work around this problem, Dror’s team designed a neural network they call ARES. (No, it’s not the Greek god of war. It’s short for Atomic Rotationally Equivariant Scorer.)
To start, the researchers trained ARES on just 18 small RNA molecules for which structures had been experimentally determined. They gave ARES these structural models specified only by their atomic structure and chemical elements.
The next test was to see if ARES could determine from this small training set the best structural model for RNA sequences it had never seen before. The researchers put it to the test with RNA molecules whose structures had been determined more recently.
ARES, however, doesn’t come up with the structures itself. Instead, the researchers give ARES a sequence and at least 1,500 possible 3D structures it might take, all generated using another computer program. Based on patterns in the training set, ARES scores each of the possible structures to find the one it predicts is closest to the actual structure. Remarkably, it does this without being provided any prior information about features important for determining RNA shapes, such as nucleotides, steric constraints, and hydrogen bonds.
It turns out that ARES consistently outperforms humans and all other previous methods to produce the best results. In fact, it outperformed at least nine other methods to come out on top in a community-wide RNA-puzzles contest. It also can make predictions about RNA molecules that are significantly larger and more complex than those upon which it was trained.
The success of ARES and this deep learning approach will help to elucidate RNA molecules with potentially important implications for health and disease. It’s another compelling example of how deep learning promises to solve many other problems in structural biology, chemistry, and the material sciences when—at the outset—very little is known.
Reference:
[1] Geometric deep learning of RNA structure. Townshend RJL, Eismann S, Watkins AM, Rangan R, Karelina M, Das R, Dror RO. Science. 2021 Aug 27;373(6558):1047-1051.
Links:
Structural Biology (National Institute of General Medical Sciences/NIH)
The Structures of Life (National Institute of General Medical Sciences/NIH)
RNA Biology (NIH)
Dror Lab (Stanford University, Palo Alto, CA)
NIH Support: National Cancer Institute; National Institute of General Medical Sciences
First Molecular Profiles of Severe COVID-19 Infections
Posted on by Dr. Francis Collins

To ensure that people with coronavirus disease 2019 (COVID-19) get the care they need, it would help if a simple blood test could predict early on which patients are most likely to progress to severe and life-threatening illness—and which are more likely to recover without much need for medical intervention. Now, researchers have provided some of the first evidence that such a test might be possible.
This tantalizing possibility comes from a study reported recently in the journal Cell. In this study, researchers took blood samples from people with mild to severe COVID-19 and analyzed them for nearly 2,000 proteins and metabolites [1]. Their detailed analyses turned up hundreds of molecular changes in blood that differentiated milder COVID-19 symptoms from more severe illness. What’s more, they found that they could train a computer to use the most informative of the proteins and predict the disease severity with a high degree of accuracy.
The findings come from the lab of Tiannan Guo, Westlake University, Zhejiang Province, China. His team recognized that, while we’ve learned a lot about the clinical symptoms of COVID-19 and the spread of the illness around the world, much less is known about the condition’s underlying molecular features. It also remains mysterious what distinguishes the 80 percent of symptomatic infected people who recover with little to no need for medical care from the other 20 percent, who suffer from much more serious illness, including respiratory distress requiring oxygen or even more significant medical interventions.
In search of clues, Guo and colleagues analyzed hundreds of molecular changes in blood samples collected from 53 healthy people and 46 people with COVID-19, including 21 with severe disease involving respiratory distress and decreased blood-oxygen levels. Their studies turned up more than 470 proteins and metabolites that differed in people with COVID-19 compared to healthy people. Of those, levels of about 300 were associated with disease severity.
Further analysis revealed that the majority of proteins and metabolites on the list are associated with the suppression or dysregulation of one of three biological processes. Two processes are related to the immune system, including early immune responses and the function of particular scavenging immune cells called macrophages. The third relates to the function of platelets, which are sticky, disc-shaped cell fragments that play an essential role in blood clotting. Such biological insights might help pave the way for potentially effective new ways to treat COVID-19 down the road.
Next, the researchers turned to “machine learning” to explore the possibility that such molecular changes also might be used to predict mild versus severe COVID-19. Machine learning involves the use of computers to discern patterns, or molecular signatures, in large data sets that a human being couldn’t readily pick out. In this case, the question was whether the computer could “learn” to tell the difference between mild and severe COVID-19 based on molecular data alone.
Their analyses showed that a computer, once trained, could differentiate mild and severe COVID-19 based on just 22 proteins and 7 metabolites. Their model correctly classified all but one person in the original training set, for an accuracy of about 94 percent. And importantly, in further prospective validation tests, they confirmed that this model accurately identified mild versus severe COVID-19 in most cases.
While these findings are certainly encouraging, there’s much more work to do. It will be important to explore these molecular signatures in many more people. It also will be critical to find out how early in the course of the disease such telltale signatures arise. While we await those answers, I find encouragement in all that we’re learning—and will continue to learn—about COVID-19 each day.
Reference:
[1] Proteomic and metabolomic characterization of COVID-19 patient sera. Shen B et al. Cell. 28 May 2020. [Epub ahead of publication]
Links:
Coronavirus (COVID-19) (NIH)
Blood Tests (National Heart, Lung, and Blood Institute/NIH)
Tiannan Guo Lab (Westlake University, Zhejiang Province, China)
Next Page