NIH Clinical Center
NIH Welcomes Visitors from OSTP
Posted on by Lawrence Tabak, D.D.S., Ph.D.

Visit the New NIH Virtual Tour
Posted on by Lawrence Tabak, D.D.S., Ph.D.
Happy Fourth of July! Before everyone heads out to celebrate the holiday with their family and friends, I want to share this brief video with you. It’s an introduction to the brand-new NIH Virtual Tour that’s now available on our website. When time permits, I encourage everyone to take the full tour of our Bethesda, MD, main campus and explore this great institution of science, technological innovation, and, above all, hope.
Among the virtual tour’s many features is an interactive, aerial map of the 32 buildings on our Bethesda campus. By clicking on a highlighted building, you can explore an impressive multimedia gallery of photos, video clips, and other resources. The tour will allow you to learn more about NIH and the ways in which we help people live longer and healthier lives.
You also can learn more about NIH’s 27 Institutes and Centers, including the NIH Clinical Center and 20 other in-depth tour stops—from research labs to patient rooms—and hear directly from some of our impressive researchers, leaders, and patients. For example, you can learn about chronic pain research from a lab in the NIH Clinical Center or see the largest zebrafish facility in the world, housed in Building 6.
What I like most about the virtual tour is that it captures what makes NIH so special—the many amazing people who collaborate every day to discover ways to solve seemingly intractable research problems. I admire their commitment to follow the science wherever it may lead.
In fact, from its humble beginnings in a one-room laboratory in 1887, NIH has become the world’s largest funder of medical research, whether that’s mobilizing to combat a deadly pandemic or strategizing to help people with a rare disorder find answers.
Not only does NIH conduct groundbreaking research in its own labs and clinics, it also supports much of the medical research conducted at universities and institutions in your states and local communities. Whether in Bethesda or beyond the Beltway, this national research effort will continue to yield the needed understanding to turn discovery into better health, helping more people to flourish and lead fully productive lives, now and in the generations to come.
That’s certainly something we can all celebrate this holiday, the 247th birthday of our great nation that I’m so honored to serve. Have a great, but safe, Fourth of July, and I’ll see you back here soon to share another blog post and another story of NIH-supported research progress.
Links:
Virtual Tour (NIH)
Visitor Information (NIH)
Groundbreaking at NIH Clinical Center
Posted on by Lawrence Tabak, D.D.S., Ph.D.

Meeting with White House Fellows
Posted on by Lawrence Tabak, D.D.S., Ph.D.

Welcome to Response Team Members
Posted on by Lawrence Tabak, D.D.S., Ph.D.

A Look Back at Science’s 2022 Breakthroughs
Posted on by Lawrence Tabak, D.D.S., Ph.D.
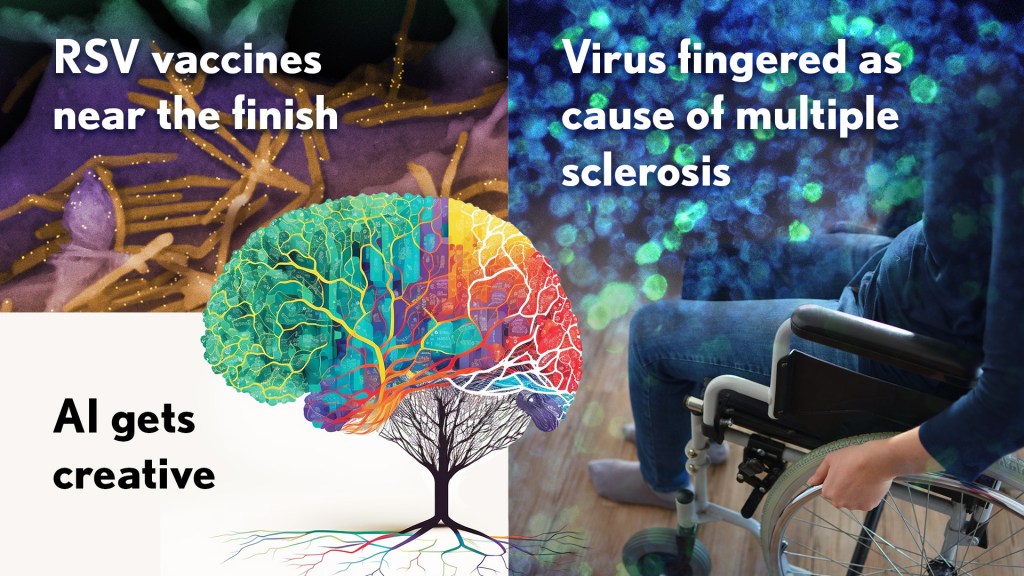
Happy New Year! I hope everyone finished 2022 with plenty to celebrate, whether it was completing a degree or certification, earning a promotion, attaining a physical fitness goal, or publishing a hard-fought scientific discovery.
If the latter, you are in good company. Last year produced some dazzling discoveries, and the news and editorial staff at the journal Science kept a watchful eye on the most high-impact advances of 2022. In December, the journal released its list of the top 10 advances across the sciences, from astronomy to zoology. In case you missed it, Science selected NASA’s James Webb Space Telescope (JWST) as the 2022 Breakthrough of the Year [1].
This unique space telescope took 20 years to complete, but it has turned out to be time well spent. Positioned 1.5-million-kilometers from Earth, the JWST and its unprecedented high-resolution images of space have unveiled the universe anew for astronomers and wowed millions across the globe checking in online. The telescope’s image stream, beyond its sheer beauty, will advance study of the early Universe, allowing astronomers to discover distant galaxies, explore the early formation of stars, and investigate the possibility of life on other planets.
While the biomedical sciences didn’t take home the top prize, they were well represented among Science’s runner-up breakthroughs. Some of these biomedical top contenders also have benefited, directly or indirectly, from NIH efforts and support. Let’s take a look:
RSV vaccines nearing the finish line: It’s been one of those challenging research marathons. But scientists last year started down the homestretch with the first safe-and-effective vaccine for respiratory syncytial virus (RSV), a leading cause of severe respiratory illness in the very young and the old.
In August, the company Pfizer presented evidence that its experimental RSV vaccine candidate offered protection for those age 60 and up. Later, they showed that the same vaccine, when administered to pregnant women, helped to protect their infants against RSV for six months after birth. Meanwhile, in October, the company GSK announced encouraging results from its late-stage phase III trial of an RSV vaccine in older adults.
As Science noted, the latest clinical progress also shows the power of basic science. For example, researchers have been working with chemically inactivated versions of the virus to develop the vaccine. But these versions have a key viral surface protein that changes its shape after fusing with a cell to start an infection. In this configuration, the protein elicits only weak levels of needed protective antibodies.
Back in 2013, Barney Graham, then with NIH’s National Institute of Allergy and Infectious Diseases (NIAID), and colleagues, solved the problem [2]. Graham’s NIH team discovered a way to lock the protein into its original prefusion state, which the immune system can better detect. This triggers higher levels of potent antibodies, and the discovery kept the science—and the marathon—moving forward.
These latest clinical advances come as RSV and other respiratory viruses, including SARS-CoV-2, the cause of COVID-19, are sending an alarming number of young children to the hospital. The hope is that researchers will cross the finish line this year or next, and we’ll have the first approved RSV vaccine.
Virus fingered as cause of multiple sclerosis: Researchers have long thought that multiple sclerosis, or MS, has a viral cause. Pointing to the right virus with the required high degree of certainty has been the challenge, slowing progress on the treatment front for those in need. As published in Science last January, Alberto Ascherio, Harvard T.H. Chan School of Public Health, Boston, and colleagues produced the strongest evidence yet that MS is caused by the Epstein-Barr virus (EBV), a herpesvirus also known for causing infectious mononucleosis [3].
The link between EBV and MS had long been suspected. But it was difficult to confirm because EBV infections are so widespread, and MS is so disproportionately rare. In the recent study, the NIH-supported researchers collected blood samples every other year from more than 10 million young adults in the U.S. military, including nearly 1,000 who were diagnosed with MS during their service. The evidence showed that the risk of an MS diagnosis increased 32-fold after EBV infection, but it held steady following infection with any other virus. Levels in blood serum of a biomarker for MS neurodegeneration also went up only after an EBV infection, suggesting that the viral illness is a leading cause for MS.
Further evidence came last year from a discovery published in the journal Nature by William Robinson, Stanford University School of Medicine, Stanford, CA, and colleagues. The NIH-supported team found a close resemblance between an EBV protein and one made in the healthy brain and spinal cord [4]. The findings suggest an EBV infection may produce antibodies that mistakenly attack the protective sheath surrounding our nerve cells. Indeed, the study showed that up to one in four people with MS had antibodies that bind both proteins.
This groundbreaking research suggests that an EBV vaccine and/or antiviral drugs that thwart this infection might ultimately prevent or perhaps even cure MS. Of note, NIAID launched last May an early-stage clinical trial for an experimental EBV vaccine at the NIH Clinical Center, Bethesda, MD.
AI Gets Creative: Science’s 2021 Breakthrough of the Year was AI-powered predictions of protein structure. In 2022, AI returned to take another well-deserved bow. This time, Science singled out AI’s now rapidly accelerating entry into once uniquely human attributes, such as artistic expression and scientific discovery.
On the scientific discovery side, Science singled out AI’s continued progress in getting creative with the design of novel proteins for vaccines and myriad other uses. One technique, called “hallucination,” generates new proteins from scratch. Researchers input random amino acid sequences into the computer, and it randomly and continuously mutates them into sequences that other AI tools are confident will fold into stable proteins. This greatly simplifies the process of protein design and frees researchers to focus their efforts on creating a protein with a desired function.
AI research now engages scientists around world, including hundreds of NIH grantees. Taking a broader view of AI, NIH recently launched the Artificial Intelligence/Machine Learning Consortium to Advance Health Equity and Researcher Diversity (AIM-AHEAD) Program. It will help to create greater diversity within the field, which is a must. A lack of diversity could perpetuate harmful biases in how AI is used, how algorithms are developed and trained, and how findings are interpreted to avoid health disparities and inequities for underrepresented communities.
And there you have it, some of the 2022 breakthroughs from Science‘s news and editorial staff. Of course, the highlighted biomedical breakthroughs don’t capture the full picture of research progress. There were many other milestone papers published in 2022 that researchers worldwide will build upon in the months and years ahead to make further progress in their disciplines and, for some, draw the attention of Science’s news and editorial staff. Here’s to another productive year in biomedical research, which the blog will continue to feature and share with you as it unfolds in 2023.
References:
[1] 2022 Breakthrough of the Year. Science. Dec 15, 2022.
[2] Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. McLellan JS, Chen M, Leung S, Kwong PD, Graham BS, et al. Science. 2013 May 31;340(6136):1113-1117.
[3] Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Bjornevik K, Cortese M, Healy BC, Kuhle J, Mina MJ, Leng Y, Elledge SJ, Niebuhr DW, Scher AI, Munger KL, Ascherio A. Science. 2022 Jan 21;375(6578):296-301.
[4] Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Lanz TV, Brewer RC, Steinman L, Robinson WH, et al. Nature. 2022 Mar;603(7900):321-327.
Links:
Respiratory Syncytial Virus (RSV) (National Institute of Allergy and Infectious Diseases/NIH)
Multiple Sclerosis (National Institute of Neurological Disorders and Stroke/NIH)
Barney Graham (Morehouse School of Medicine, Atlanta)
Alberto Ascherio (Harvard T.H. Chan School of Public Health, Boston)
Robinson Lab (Stanford Medicine, Stanford, CA)
James Webb Space Telescope (Goddard Space Flight Center/NASA, Greenbelt, MD)
Clinical Center Doctors Testing 3D-Printed Miniature Ventilator
Posted on by James K. Gilman, MD, NIH Clinical Center

Here at the NIH Clinical Center, we are proud to be considered a world-renowned research hospital that provides hope through pioneering clinical research to improve human health. But what you may not know is that our doctors are constantly partnering with public and private sectors to come up with innovative technologies that will help to advance health outcomes.
I’m excited to bring to you a story that is perfect example of the ingenuity of our NIH doctors working with global strategic partners to create potentially life-saving technologies. This story begins during the COVID-19 pandemic with the global shortage of ventilators to help patients breathe. Hospitals had a profound need for inexpensive, easy-to-use, rapidly mass-produced resuscitation devices that could be quickly distributed in areas of critical need.
Through strategic partnerships, our Clinical Center doctors learned about and joined an international group of engineers, physicians, respiratory therapists, and patient advocates using their engineering skills to create a ventilator that was functional, affordable, and intuitive. After several iterations and bench testing, they devised a user-friendly ventilator.
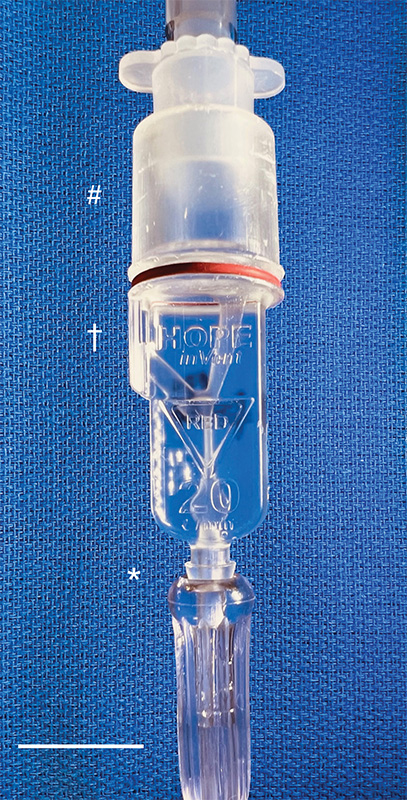
Then, with the assistance of 3D-printing technology, they improved the original design and did something pretty incredible: the team created the smallest single-patient ventilator seen to date. The device is just 2.4 centimeters (about 1 inch) in diameter with a length of 7.4 centimeters (about 3 inches).
A typical ventilator in a hospital obviously is much larger and has a bellows system. It fills with oxygen and then forces it into the lungs followed by the patient passively exhaling. These systems have multiple moving parts, valves, hoses, and electronic or mechanical controls to manage all aspects of the oxygen flow into the lungs.
But our miniature, 3D-printed ventilator is single use, disposable, and has no moving parts. It’s based on principles of fluidics to ventilate patients by automatically oscillating between forced inspiration and assisted expiration as airway pressure changes. It requires only a continuous supply of pressurized oxygen.
The possibilities of this 3D-printed miniature ventilator are broad. The ventilators could be easily used in emergency transport, potentially treating battlefield casualties or responding to disasters and mass casualty events like earthquakes.
While refining a concept is important, the key is converting it to actual use, which our doctors are doing admirably in their preclinical and clinical studies. NIH’s William Pritchard, Andrew Mannes, Brad Wood, John Karanian, Ivane Bakhutashvili, Matthew Starost, David Eckstein, and medical student Sheridan Reed studied and have already tested the ventilators in swine with acute lung injury, a common severe outcome in a number of respiratory threats including COVID-19.
In the study, the doctors tested three versions of the device built to correspond to mild, moderate, and severe lung injury. The respirators provided adequate support for moderate and mild lung injuries, and the doctors recall how amazing it was initially to witness a 190-pound swine ventilated by this miniature ventilator.
The doctors believe that the 3D-printed miniature ventilator is a potential “game changer” from start to finish since it is lifesaving, small, simple to use, can be easily and inexpensively printed and stored, and does not require additional maintenance. They recently published their preclinical trial results in the journal Science Translational Medicine [1].
The NIH team is preparing to initiate first-in-human trials here at the Clinical Center in the coming months. Perhaps, in the not-too-distant future, a device designed to help people breathe could fit into your pocket next to your phone and keys.
Reference:
[1] In-line miniature 3D-printed pressure-cycled ventilator maintains respiratory homeostasis in swine with induced acute pulmonary injury. Pritchard WF, Karanian JW, Jung C, Bakhutashvili I, Reed SL, Starost MF, Froelke BR, Barnes TR, Stevenson D, Mendoza A, Eckstein DJ, Wood BJ, Walsh BK, Mannes AJ. Sci Transl Med. 2022 Oct 12;14(666):eabm8351.
Links:
Clinical Center (NIH)
Andrew Mannes (Clinical Center)
Bradford Wood (Clinical Center)
David Eckstein (Clinical Center)
Note: Dr. Lawrence Tabak, who performs the duties of the NIH Director, has asked the heads of NIH’s Institutes and Centers (ICs) to contribute occasional guest posts to the blog to highlight some of the interesting science that they support and conduct. This is the 21st in the series of NIH IC guest posts that will run until a new permanent NIH director is in place.
Special Thanks for A Job Well Done
Posted on by Lawrence Tabak, D.D.S., Ph.D.

Laura will be especially missed. She has presided over the CCRHRB with distinction from its first inception in July 2016. Afterwards, photos were taken to mark the occasion. That includes this one showing (l-r) Tara Schwetz, NIH’s Acting Principal Deputy Director; James Gilman, CEO of the Clinical Center; Laura Forese; and me. Credit: NIH
Cutting Ribbon for NIH Clinical Center Pharmacy
Posted on by Lawrence Tabak, D.D.S., Ph.D.

I’m third from the left in the ribbon-cutting line. To my right, scissors in hand, (l-r) are Richard DeCederfelt, the Clinical Center’s Acting Pharmacy Chief, and James Gilman, CEO of the Clinical Center. Cutting the ribbon to my left (l-r) are Alfred Johnson, NIH’s Deputy Director for Management, and Marilyn Farinre, the Clinical Center’s Pharmacy Operations Chief. Looking on just behind them (l-r) are Tara Schwetz, NIH’s Acting Principal Deputy Director, and Michael Gottesman, NIH’s Deputy Director for Intramural Research. The ribbon-cutting ceremony took place on May 18 in the Outpatient Pharmacy Waiting Room. Credit: NIH
A Special Thanksgiving Day Concert
Posted on by Dr. Francis Collins

Next Page