epigenomics
Brain Atlas Paves the Way for New Understanding of How the Brain Functions
Posted on by Lawrence Tabak, D.D.S., Ph.D.

When NIH launched The BRAIN Initiative® a decade ago, one of many ambitious goals was to develop innovative technologies for profiling single cells to create an open-access reference atlas cataloguing the human brain’s many parts. The ultimate goal wasn’t to produce a single, static reference map, but rather to capture a dynamic view of how the brain’s many cells of varied types are wired to work together in the healthy brain and how this picture may shift in those with neurological and mental health disorders.
So I’m now thrilled to report the publication of an impressive collection of work from hundreds of scientists in the BRAIN Initiative Cell Census Network (BICCN), detailed in more than 20 papers in Science, Science Advances, and Science Translational Medicine.1 Among many revelations, this unprecedented, international effort has characterized more than 3,000 human brain cell types. To put this into some perspective, consider that the human lung contains 61 cell types.2 The work has also begun to uncover normal variation in the brains of individual people, some of the features that distinguish various disease states, and distinctions among key parts of the human brain and those of our closely related primate cousins.
Of course, it’s not possible to do justice to this remarkable body of work or its many implications in the space of a single blog post. But to give you an idea of what’s been accomplished, some of these studies detail the primary effort to produce a comprehensive brain atlas, including defining the brain’s many cell types along with their underlying gene activity and the chemical modifications that turn gene activity up or down.3,4,5
Other studies in this collection take a deep dive into more specific brain areas. For instance, to capture normal variations among people, a team including Nelson Johansen, University of California, Davis, profiled cells in the neocortex—the outermost portion of the brain that’s responsible for many complex human behaviors.6 Overall, the work revealed a highly consistent cellular makeup from one person to the next. But it also highlighted considerable variation in gene activity, some of which could be explained by differences in age, sex and health. However, much of the observed variation remains unexplained, opening the door to more investigations to understand the meaning behind such brain differences and their role in making each of us who we are.
Yang Li, now at Washington University in St. Louis, and his colleagues analyzed 1.1 million cells from 42 distinct brain areas in samples from three adults.4 They explored various cell types with potentially important roles in neuropsychiatric disorders and were able to pinpoint specific cell types, genes and genetic switches that may contribute to the development of certain traits and disorders, including bipolar disorder, depression and schizophrenia.
Yet another report by Nikolas Jorstad, Allen Institute, Seattle, and colleagues delves into essential questions about what makes us human as compared to other primates like chimpanzees.7 Their comparisons of gene activity at the single-cell level in a specific area of the brain show that humans and other primates have largely the same brain cell types, but genes are activated differently in specific cell types in humans as compared to other primates. Those differentially expressed genes in humans often were found in portions of the genome that show evidence of rapid change over evolutionary time, suggesting that they play important roles in human brain function in ways that have yet to be fully explained.
All the data represented in this work has been made publicly accessible online for further study. Meanwhile, the effort to build a more finely detailed picture of even more brain cell types and, with it, a more complete understanding of human brain circuitry and how it can go awry continues in the BRAIN Initiative Cell Atlas Network (BICAN). As impressive as this latest installment is—in our quest to understand the human brain, brain disorders, and their treatment—we have much to look forward to in the years ahead.
References:
A list of all the papers part of the brain atlas research is available here: https://www.science.org/collections/brain-cell-census.
[1] M Maroso. A quest into the human brain. Science DOI: 10.1126/science.adl0913 (2023).
[2] L Sikkema, et al. An integrated cell atlas of the lung in health and disease. Nature Medicine DOI: 10.1038/s41591-023-02327-2 (2023).
[3] K Siletti, et al. Transcriptomic diversity of cell types across the adult human brain. Science DOI: 10.1126/science.add7046 (2023).
[4] Y Li, et al. A comparative atlas of single-cell chromatin accessibility in the human brain. Science DOI: 10.1126/science.adf7044 (2023).
[5] W Tian, et al. Single-cell DNA methylation and 3D genome architecture in the human brain. Science DOI: 10.1126/science.adf5357 (2023).
[6] N Johansen, et al. Interindividual variation in human cortical cell type abundance and expression. Science DOI: 10.1126/science.adf2359 (2023).
[7] NL Jorstad, et al. Comparative transcriptomics reveals human-specific cortical features. Science DOI: 10.1126/science.ade9516 (2023).
NIH Support: Projects funded through the NIH BRAIN Initiative Cell Consensus Network
Taking a Deep Dive into the Alzheimer’s Brain in Search of Understanding and New Targets
Posted on by Lawrence Tabak, D.D.S., Ph.D.

People living with Alzheimer’s disease experience a gradual erosion of memory and thinking skills until they can no longer carry out daily activities. Hallmarks of the disease include the buildup of plaques that collect between neurons, accumulations of tau protein inside neurons and weakening of neural connections. However, there’s still much to learn about what precisely happens in the Alzheimer’s brain and how the disorder’s devastating march might be slowed or even stopped. Alzheimer’s affects more than six million people in the United States and is the seventh leading cause of death among adults in the U.S., according to the National Institute on Aging.
NIH-supported researchers recently published a trove of data in the journal Cell detailing the molecular drivers of Alzheimer’s disease and which cell types in the brain are most likely to be affected.1,2,3,4 The scientists, led by Li-Huei Tsai and Manolis Kellis, both at the Massachusetts Institute of Technology, Cambridge, MA, characterized gene activity at the single-cell level in more than two million cells from postmortem brain tissue. They also assessed DNA damage and surveyed epigenetic changes in cells, which refers to chemical modifications to DNA that alter gene expression in the cell. The findings could help researchers pinpoint new targets for Alzheimer’s disease treatments.
In the first of four studies, the researchers examined 54 brain cell types in 427 brain samples from a cohort of people with varying levels of cognitive impairment that has been followed since 1994.1 The MIT team generated an atlas of gene activity patterns within the brain’s prefrontal cortex, an important area for memory retrieval.
Their analyses in brain samples taken from people with Alzheimer’s dementia showed altered activity in genes involved in various functions. Additional findings showed that people with normal cognitive abilities with evidence of plaques in their brains had more neurons that inhibit or dampen activity in the prefrontal cortex compared to those with Alzheimer’s dementia. The finding suggests that the workings of inhibitory neurons may play an unexpectedly important role in maintaining cognitive resilience despite age-related changes, including the buildup of plaques. It’s one among many discoveries that now warrant further study.
In another report, the researchers compared brain tissues from 48 people without Alzheimer’s to 44 people with early- or late-stage Alzheimer’s.2 They developed a map of the various elements that regulate function within cells in the prefrontal cortex. By cross-referencing epigenomic and gene activity data, the researchers showed changes in many genes with known links to Alzheimer’s disease development and risk.
Their single-cell analysis also showed that these changes most often occur in microglia, which are immune cells that remove cellular waste products from the brain. At the same time, every cell type they studied showed a breakdown over time in the cells’ normal epigenomic patterning, a process that may cause a cell to behave differently as it loses essential aspects of its original identity and function.
In a third report, the researchers looked even deeper into gene activity within the brain’s waste-clearing microglia.3 Based on the activity of hundreds of genes, they were able to define a dozen distinct microglia “states.” They also showed that more microglia enter an inflammatory state in the Alzheimer’s brain compared to a healthy human brain. Fewer microglia in the Alzheimer’s brain were in a healthy, balanced state as well. The findings suggest that treatments that target microglia to reduce inflammation and promote balance may hold promise for treating Alzheimer’s disease.
The fourth and final report zeroed in on DNA damage, inspired in part by earlier findings suggesting greater damage within neurons even before Alzheimer’s symptoms appear.4 In fact, breaks in DNA occur as part of the normal process of forming new memories. But those breaks in the healthy brain are quickly repaired as the brain makes new connections.
The researchers studied postmortem brain tissue samples and found that, over time in the Alzheimer’s brain, the damage exceeds the brain’s ability to repair it. As a result, attempts to put the DNA back together leads to a patchwork of mistakes, including rearrangements in the DNA and fusions as separate genes are merged. Such changes appear to arise especially in genes that control neural connections, which may contribute to the signs and symptoms of Alzheimer’s.
The researchers say they now plan to apply artificial intelligence and other analytic tools to learn even more about Alzheimer’s disease from this trove of data. To speed progress even more, they’ve made all the data freely available online to the research community, where it promises to yield many more fundamentally important discoveries about the precise underpinnings of Alzheimer’s disease in the brain and new ways to intervene in Alzheimer’s dementia.
References:
[1] Mathys H, et al. Single-cell atlas reveals correlates of high cognitive function, dementia, and resilience to Alzheimer’s disease pathology. Cell. DOI: 10.1016/j.cell.2023.08.039. (2023).
[2] Xiong X, et al. Epigenomic dissection of Alzheimer’s disease pinpoints causal variants and reveals epigenome erosion. Cell. DOI: 10.1016/j.cell.2023.08.040. (2023).
[3] Sun N, et al. Human microglial state dynamics in Alzheimer’s disease progression. Cell. DOIi: 10.1016/j.cell.2023.08.037. (2023).
[4] Dileep V, et al. Neuronal DNA double-strand breaks lead to genome structural variations and 3D genome disruption in neurodegeneration. Cell. 2023 DOI: 10.1016/j.cell.2023.08.038. (2023).
NIH Support: National Institute on Aging, National Institute of Neurological Disorders and Stroke, National Institute of Mental Health, National Institute of General Medical Sciences
Mood-Altering Messenger Goes Nuclear
Posted on by Dr. Francis Collins
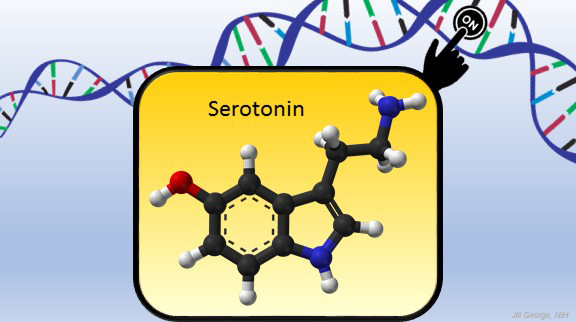
Serotonin is best known for its role as a chemical messenger in the brain, helping to regulate mood, appetite, sleep, and many other functions. It exerts these influences by binding to its receptor on the surface of neural cells. But startling new work suggests the impact of serotonin does not end there: the molecule also can enter a cell’s nucleus and directly switch on genes.
While much more study is needed, this is a potentially groundbreaking discovery. Not only could it have implications for managing depression and other mood disorders, it may also open new avenues for treating substance abuse and neurodegenerative diseases.
To understand how serotonin contributes to switching genes on and off, a lesson on epigenetics is helpful. Keep in mind that the DNA instruction book of all cells is essentially the same, yet the chapters of the book are read in very different ways by cells in different parts of the body. Epigenetics refers to chemical marks on DNA itself or on the protein “spools” called histones that package DNA. These marks influence the activity of genes in a particular cell without changing the underlying DNA sequence, switching them on and off or acting as “volume knobs” to turn the activity of particular genes up or down.
The marks include various chemical groups—including acetyl, phosphate, or methyl—which are added at precise locations to those spool-like proteins called histones. The addition of such groups alters the accessibility of the DNA for copying into messenger RNA and producing needed proteins.
In the study reported in Nature, researchers led by Ian Maze and postdoctoral researcher Lorna Farrelly, Icahn School of Medicine at Mount Sinai, New York, followed a hunch that serotonin molecules might also get added to histones [1]. There had been hints that it might be possible. For instance, earlier evidence suggested that inside cells, serotonin could enter the nucleus. There also was evidence that serotonin could attach to proteins outside the nucleus in a process called serotonylation.
These data begged the question: Is serotonylation important in the brain and/or other living tissues that produce serotonin in vivo? After a lot of hard work, the answer now appears to be yes.
These NIH-supported researchers found that serotonylation does indeed occur in the cell nucleus. They also identified a particular enzyme that directly attaches serotonin molecules to histone proteins. With serotonin attached, DNA loosens on its spool, allowing for increased gene expression.
The team found that histone serotonylation takes place in serotonin-producing human neurons derived from induced pluripotent stem cells (iPSCs). They also observed this process occurring in the brains of developing mice.
In fact, the researchers found evidence of those serotonin marks in many parts of the body. They are especially prevalent in the brain and gut, where serotonin also is produced in significant amounts. Those marks consistently correlate with areas of active gene expression.
The serotonin mark often occurs on histones in combination with a second methyl mark. The researchers suggest that this double marking of histones might help to further reinforce an active state of gene expression.
This work demonstrates that serotonin can directly influence gene expression in a manner that’s wholly separate from its previously known role in transmitting chemical messages from one neuron to the next. And, there are likely other surprises in store.
The newly discovered role of serotonin in modifying gene expression may contribute significantly to our understanding of mood disorders and other psychiatric conditions with known links to serotonin signals, suggesting potentially new targets for therapeutic intervention. But for now, this fundamental discovery raises many more intriguing questions than it answers.
Science is full of surprises, and this paper is definitely one of them. Will this kind of histone marking occur with other chemical messengers, such as dopamine and acetylcholine? This unexpected discovery now allows us to track serotonin and perhaps some of the brain’s other chemical messengers to see what they might be doing in the cell nucleus and whether this information might one day help in treating the millions of Americans with mood and behavioral disorders.
Reference:
[1] Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Farrelly LA, Thompson RE, Zhao S, Lepack AE, Lyu Y, Bhanu NV, Zhang B, Loh YE, Ramakrishnan A, Vadodaria KC, Heard KJ, Erikson G, Nakadai T, Bastle RM, Lukasak BJ, Zebroski H 3rd, Alenina N, Bader M, Berton O, Roeder RG, Molina H, Gage FH, Shen L, Garcia BA, Li H, Muir TW, Maze I. Nature. 2019 Mar 13. [Epub ahead of print]
Links:
Any Mood Disorder (National Institute of Mental Health/NIH)
Drugs, Brains, and Behavior: The Science of Addiction (National Institute on Drug Abuse/NIH)
Epigenomics (National Human Genome Research Institute/NIH)
Maze Lab (Icahn School of Medicine at Mount Sinai, New York, NY)
NIH Support: National Institute on Drug Abuse; National Institute of Mental Health; National Institute of General Medical Sciences; National Cancer Institute
Creative Minds: A New Way to Look at Cancer
Posted on by Dr. Francis Collins

Bradley Bernstein
Inside our cells, strands of DNA wrap around spool-like histone proteins to form a DNA-histone complex called chromatin. Bradley Bernstein, a pathologist at Massachusetts General Hospital, Harvard University, and Broad Institute, has always been fascinated by this process. What interests him is the fact that an approximately 6-foot-long strand of DNA can be folded and packed into orderly chromatin structures inside a cell nucleus that’s just 0.0002 inch wide.
Bernstein’s fascination with DNA packaging led to the recent major discovery that, when chromatin misfolds in brain cells, it can activate a gene associated with the cancer glioma [1]. This suggested a new cancer-causing mechanism that does not require specific DNA mutations. Now, with a 2016 NIH Director’s Pioneer Award, Bernstein is taking a closer look at how misfolded and unstable chromatin can drive tumor formation, and what that means for treating cancer.
Creative Minds: Using Machine Learning to Understand Genome Function
Posted on by Dr. Francis Collins

Science has always fascinated Anshul Kundaje, whether it was biology, physics, or chemistry. When he left his home country of India to pursue graduate studies in electrical engineering at Columbia University, New York, his plan was to focus on telecommunications and computer networks. But a course in computational genomics during his first semester showed him he could follow his interest in computing without giving up his love for biology.
Now an assistant professor of genetics and computer science at Stanford University, Palo Alto, CA, Kundaje has received a 2016 NIH Director’s New Innovator Award to explore not just how the human genome sequence encodes function, but also why it functions in the way that it does. Kundaje even envisions a time when it might be possible to use sophisticated computational approaches to predict the genomic basis of many human diseases.
Creative Minds: Building the RNA Toolbox
Posted on by Dr. Francis Collins

Caption: Genetically identical mice. The Agouti gene is active in the yellow mouse and inactive in the brown mouse.
Credit: Dana Dolinoy, University of Michigan, Ann Arbor, and Randy Jirtle, Duke University, Durham, NC
Step inside the lab of Dana Dolinoy at the University of Michigan, Ann Arbor, and you’re sure to hear conversations that include the rather strange word “agouti” (uh-goo-tee). In this context, it’s a name given to a strain of laboratory mice that arose decades ago from a random mutation in the Agouti gene, which is normally expressed only transiently in hair follicles. The mutation causes the gene to be turned on, or expressed, continuously in all cell types, producing mice that are yellow, obese, and unusually prone to developing diabetes and cancer. As it turns out, these mutant mice and the gene they have pointed to are more valuable than ever today because they offer Dolinoy and other researchers an excellent model for studying the rapidly emerging field of epigenomics.
The genome of the mouse, just as for the human, is the complete DNA instruction book; it contains the coding information for building the proteins that carry out a variety of functions in a cell. But modifications to the DNA determine its function, and these are collectively referred to as the epigenome. The epigenome is made up of chemical tags and proteins that can attach to the DNA and direct such actions as turning genes on or off, thereby controlling the production of proteins in particular cells. These tags have different patterns in each cell type, helping to explain, for example, why a kidney and a skin cell can behave so differently when they share the same DNA.
Some types of genes, including Agouti, are particularly vulnerable to epigenomic effects. In fact, Dolinoy has discovered that exposing normal, wild-type (brown) mice to certain chemicals and dietary factors during pregnancy can switch on the Agouti gene in their developing offspring, turning their coats yellow and their health poor. Dolinoy says these experiments raise much larger questions: If researchers discover populations of humans that have been exposed to lifestyle or environmental factors that modify their epigenomes in ways that may possibly contribute to risk for certain diseases, can the modification be passed on to their children and grandchildren (referred to as transgenerational epigenetic inheritance, a controversial topic)? If so, how can we develop the high-precision tools needed to better understand and perhaps even reduce such risks? The University of Michigan researcher received a 2015 NIH Director’s Transformative Research Award to undertake that challenge.
Flipping a Genetic Switch on Obesity?
Posted on by Dr. Francis Collins
 When weight loss is the goal, the equation seems simple enough: consume fewer calories and burn more of them exercising. But for some people, losing and keeping off the weight is much more difficult for reasons that can include a genetic component. While there are rare genetic causes of extreme obesity, the strongest common genetic contributor discovered so far is a variant found in an intron of the FTO gene. Variations in this untranslated region of the gene have been tied to differences in body mass and a risk of obesity [1]. For the one in six people of European descent born with two copies of the risk variant, the consequence is carrying around an average of an extra 7 pounds [2].
When weight loss is the goal, the equation seems simple enough: consume fewer calories and burn more of them exercising. But for some people, losing and keeping off the weight is much more difficult for reasons that can include a genetic component. While there are rare genetic causes of extreme obesity, the strongest common genetic contributor discovered so far is a variant found in an intron of the FTO gene. Variations in this untranslated region of the gene have been tied to differences in body mass and a risk of obesity [1]. For the one in six people of European descent born with two copies of the risk variant, the consequence is carrying around an average of an extra 7 pounds [2].
Now, NIH-funded researchers reporting in The New England Journal of Medicine [3] have figured out how this gene influences body weight. The answer is not, as many had suspected, in regions of the brain that control appetite, but in the progenitor cells that produce white and beige fat. The researchers found that the risk variant is part of a larger genetic circuit that determines whether our bodies burn or store fat. This discovery may yield new approaches to intervene in obesity with treatments designed to change the way fat cells handle calories.
What Makes Our Brain Human? The Search for Answers
Posted on by Dr. Francis Collins

“The Thinker” by Auguste Rodin (photo by Brian Hillegas)
Humans’ most unique traits, such as speaking and abstract thinking, are rooted in the outer layer of our brains called the cerebral cortex. This convoluted sheet of grey matter is found in all mammals, but it is much larger and far more complex in Homo sapiens than in any other species. The cortex comprises nearly 80 percent of our brain mass, with some 16 billion neurons packed into more than 50 distinct, meticulously organized regions.
In an effort to explore the evolution of the human cortex, many researchers have looked to changes in the portion of the genome that codes for proteins. But a new paper, published in the journal Science [1], shows that protein-coding DNA provides only part of the answer. The new findings reveal that an even more critical component may be changes in the DNA sequences that regulate the activity of these genes.
NIH Common Fund: 10 Years of Transformative Science
Posted on by Dr. Francis Collins
 Happy 10th Anniversary to the Common Fund! It’s hard to believe that it’s been a decade since I joined then-NIH Director Elias Zerhouni at the National Press Club to launch this trans-NIH effort to catalyze innovation and speed progress across many fields of biomedical research.
Happy 10th Anniversary to the Common Fund! It’s hard to believe that it’s been a decade since I joined then-NIH Director Elias Zerhouni at the National Press Club to launch this trans-NIH effort to catalyze innovation and speed progress across many fields of biomedical research.
We’re marking this milestone with a special celebration today at NIH’s main campus. And, for those of you who can’t make it to Bethesda to join in the festivities, you can watch the videocast (live or archived). But allow me also to take this opportunity to share just a bit of the history and a few of the many achievements of this bold new approach to the support of science.
Creative Minds: Interpreting Your Genome
Posted on by Dr. Francis Collins

Credit: Jane Ades, National Human Genome Research Institute, NIH
Just this year, we’ve reached the point where we can sequence an entire human genome for less than $1,000. That’s great news—and rather astounding, since the first human genome sequence (finished in 2003) cost an estimated $400,000,000! Does that mean we’ll be able to use each person’s unique genetic blueprint to guide his or her health care from cradle to grave? Maybe eventually, but it’s not quite as simple as it sounds.
Before we can use your genome to develop more personalized strategies for detecting, treating, and preventing disease, we need to be able to interpret the many variations that make your genome distinct from everybody else’s. While most of these variations are neither bad nor good, some raise the risk of particular diseases, and others serve to lower the risk. How do we figure out which is which?
Jay Shendure, an associate professor at the University of Washington in Seattle, has an audacious plan to figure this out, which is why he is among the 2013 recipients of the NIH Director’s Pioneer Award.