variants
How COVID-19 Immunity Holds Up Over Time
Posted on by Lawrence Tabak, D.D.S., Ph.D.
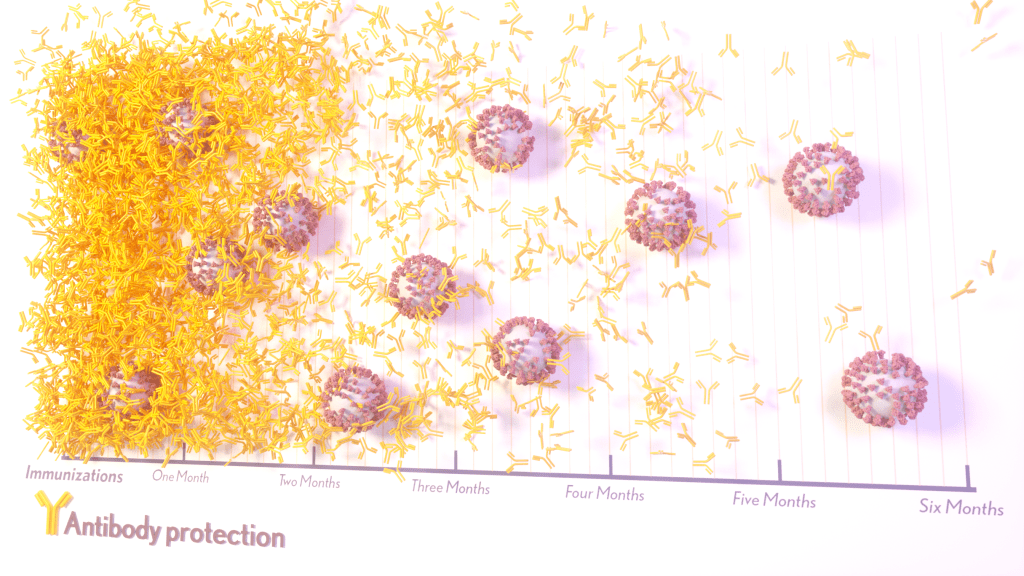
More than 215 million people in the United States are now fully vaccinated against the SARS-CoV-2 virus responsible for COVID-19 [1]. More than 40 percent—more than 94 million people—also have rolled up their sleeves for an additional, booster dose. Now, an NIH-funded study exploring how mRNA vaccines are performing over time comes as a reminder of just how important it will be to keep those COVID-19 vaccines up to date as coronavirus variants continue to circulate.
The results, published in the journal Science Translational Medicine, show that people who received two doses of either the Pfizer or Moderna COVID-19 mRNA vaccines did generate needed virus-neutralizing antibodies [2]. But levels of those antibodies dropped considerably after six months, suggesting declining immunity over time.
The data also reveal that study participants had much reduced protection against newer SARS-CoV-2 variants, including Delta and Omicron. While antibody protection remained stronger in people who’d also had a breakthrough infection, even that didn’t appear to offer much protection against infection by the Omicron variant.
The new study comes from a team led by Shan-Lu Liu at The Ohio State University, Columbus. They wanted to explore how well vaccine-acquired immune protection holds up over time, especially in light of newly arising SARS-CoV-2 variants.
This is an important issue going forward because mRNA vaccines train the immune system to produce antibodies against the spike proteins that crown the surface of the SARS-CoV-2 coronavirus. These new variants often have mutated, or slightly changed, spike proteins compared to the original one the immune system has been trained to detect, potentially dampening the immune response.
In the study, the team collected serum samples from 48 fully vaccinated health care workers at four key time points: 1) before vaccination, 2) three weeks after the first dose, 3) one month after the second dose, and 4) six months after the second dose.
They then tested the ability of antibodies in those samples to neutralize spike proteins as a correlate for how well a vaccine works to prevent infection. The spike proteins represented five major SARS-CoV-2 variants. The variants included D614G, which arose very soon after the coronavirus first was identified in Wuhan and quickly took over, as well as Alpha (B.1.1.7), Beta (B.1.351), Delta (B.1.617.2), and Omicron (B.1.1.529).
The researchers explored in the lab how neutralizing antibodies within those serum samples reacted to SARS-CoV-2 pseudoviruses representing each of the five variants. SARS-CoV-2 pseudoviruses are harmless viruses engineered, in this case, to bear coronavirus spike proteins on their surfaces. Because they don’t replicate, they are safe to study without specially designed biosafety facilities.
At any of the four time points, antibodies showed a minimal ability to neutralize the Omicron spike protein, which harbors about 30 mutations. These findings are consistent with an earlier study showing a significant decline in neutralizing antibodies against Omicron in people who’ve received the initial series of two shots, with improved neutralizing ability following an additional booster dose.
The neutralizing ability of antibodies against all other spike variants showed a dramatic decline from 1 to 6 months after the second dose. While there was a marked decline over time after both vaccines, samples from health care workers who’d received the Moderna vaccine showed about twice the neutralizing ability of those who’d received the Pfizer vaccine. The data also suggests greater immune protection in fully vaccinated healthcare workers who’d had a breakthrough infection with SARS-CoV-2.
In addition to recommending full vaccination for all eligible individuals, the Centers for Disease Control and Prevention (CDC) now recommends everyone 12 years and up should get a booster dose of either the Pfizer or Moderna vaccines at least five months after completing the primary series of two shots [3]. Those who’ve received the Johnson & Johnson vaccine should get a booster at least two months after receiving the initial dose.
While plenty of questions about the durability of COVID-19 immunity over time remain, it’s clear that the rapid deployment of multiple vaccines over the course of this pandemic already has saved many lives and kept many more people out of the hospital. As the Omicron threat subsides and we start to look forward to better days ahead, it will remain critical for researchers and policymakers to continually evaluate and revise vaccination strategies and recommendations, to keep our defenses up as this virus continues to evolve.
References:
[1] COVID-19 vaccinations in the United States. Centers for Disease Control and Prevention. February 27, 2022.
[2] Neutralizing antibody responses elicited by SARS-CoV-2 mRNA vaccination wane over time and are boosted by breakthrough infection. Evans JP, Zeng C, Carlin C, Lozanski G, Saif LJ, Oltz EM, Gumina RJ, Liu SL. Sci Transl Med. 2022 Feb 15:eabn8057.
[3] COVID-19 vaccine booster shots. Centers for Disease Control and Prevention. Feb 2, 2022.
Links:
COVID-19 Research (NIH)
Shan-Lu Liu (The Ohio State University, Columbus)
NIH Support: National Institute of Allergy and Infectious Diseases; National Cancer Institute; National Heart, Lung, and Blood Institute; Eunice Kennedy Shriver National Institute of Child Health and Human Development
Latest on Omicron Variant and COVID-19 Vaccine Protection
Posted on by Dr. Francis Collins
There’s been great concern about the new Omicron variant of SARS-CoV-2, the coronavirus that causes COVID-19. A major reason is Omicron has accumulated over 50 mutations, including about 30 in the spike protein, the part of the coronavirus that mRNA vaccines teach our immune systems to attack. All of these genetic changes raise the possibility that Omicron could cause breakthrough infections in people who’ve already received a Pfizer or Moderna mRNA vaccine.
So, what does the science show? The first data to emerge present somewhat encouraging results. While our existing mRNA vaccines still offer some protection against Omicron, there appears to be a significant decline in neutralizing antibodies against this variant in people who have received two shots of an mRNA vaccine.
However, initial results of studies conducted both in the lab and in the real world show that people who get a booster shot, or third dose of vaccine, may be better protected. Though these data are preliminary, they suggest that getting a booster will help protect people already vaccinated from breakthrough or possible severe infections with Omicron during the winter months.
Though Omicron was discovered in South Africa only last month, researchers have been working around the clock to learn more about this variant. Last week brought the first wave of scientific data on Omicron, including interesting work from a research team led by Alex Sigal, Africa Health Research Institute, Durban, South Africa [1].
In lab studies working with live Omicron virus, the researchers showed that this variant still relies on the ACE2 receptor to infect human lung cells. That’s really good news. It means that the therapeutic tools already developed, including vaccines, should generally remain useful for combatting this new variant.
Sigal and colleagues also tested the ability of antibodies in the plasma from 12 fully vaccinated individuals to neutralize Omicron. Six of the individuals had no history of COVID-19. The other six had been infected with the original variant in the first wave of infections in South Africa.
As expected, the samples showed very strong neutralization against the original SARS-CoV-2 variant. However, antibodies from people who’d been previously vaccinated with the two-dose Pfizer vaccine took a significant hit against Omicron, showing about a 40-fold decline in neutralizing ability.
This escape from immunity wasn’t complete. Indeed, blood samples from five individuals showed relatively good antibody levels against Omicron. All five had previously been infected with SARS-CoV-2 in addition to being vaccinated. These findings add to evidence on the value of full vaccination for protecting against reinfections in people who’ve had COVID-19 previously.
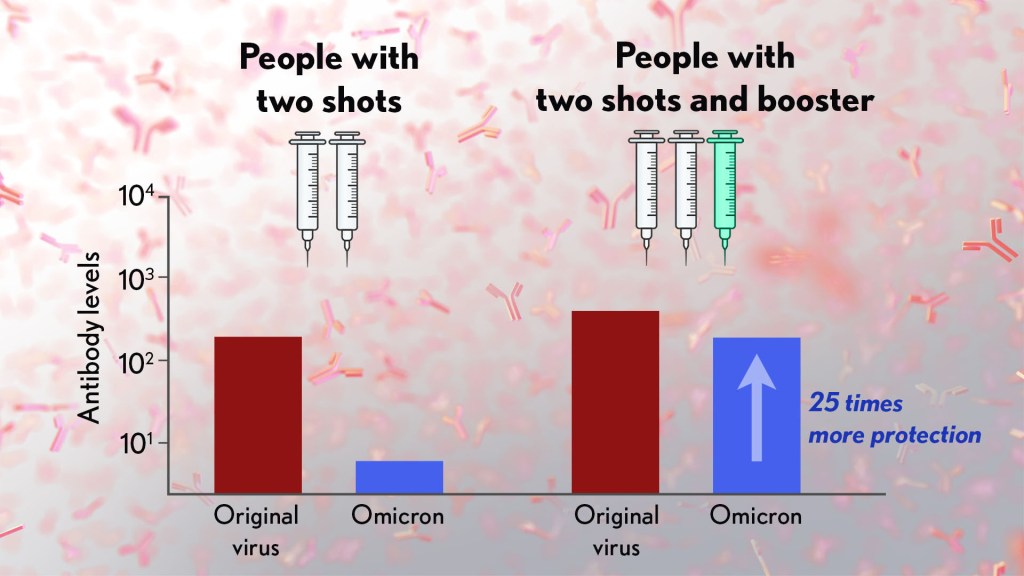
Also of great interest were the first results of the Pfizer study, which the company made available in a news release [2]. Pfizer researchers also conducted laboratory studies to test the neutralizing ability of blood samples from 19 individuals one month after a second shot compared to 20 others one month after a booster shot.
These studies showed that the neutralizing ability of samples from those who’d received two shots had a more than 25-fold decline relative to the original virus. Together with the South Africa data, it suggests that the two-dose series may not be enough to protect against breakthrough infections with the Omicron variant.
In much more encouraging news, their studies went on to show that a booster dose of the Pfizer vaccine raised antibody levels against Omicron to a level comparable to the two-dose regimen against the original variant (as shown in the figure above). While efforts already are underway to develop an Omicron-specific COVID-19 vaccine, these findings suggest that it’s already possible to get good protection against this new variant by getting a booster shot.
Very recently, real-world data from the United Kingdom, where Omicron cases are rising rapidly, are providing additional evidence for how boosters can help. In a preprint [3], Andrews et. al showed the effectiveness of two shots of Pfizer mRNA vaccine trended down after four months to about 40 percent. That’s not great, but note that 40 percent is far better than zero. So, clearly there is some protection provided.
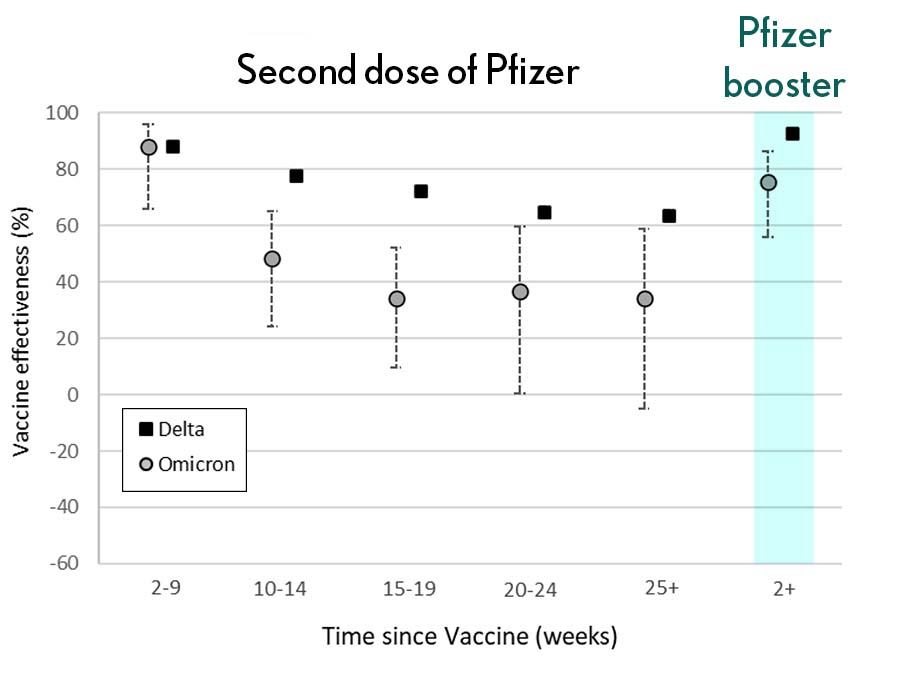
Most impressively (as shown in the figure from Andrews N, et al.) a booster substantially raised that vaccine effectiveness to about 80 percent. That’s not quite as high as for Delta, but certainly an encouraging result. Once again, these data show that boosting the immune system after a pause produces enhanced immunity against new viral variants, even though the booster was designed from the original virus. Your immune system is awfully clever. You get both quantitative and qualitative benefits.
It’s also worth noting that the Omicron variant mostly doesn’t have mutations in portions of its genome that are the targets of other aspects of vaccine-induced immunity, including T cells. These cells are part of the body’s second line of defense and are generally harder for viruses to escape. While T cells can’t prevent infection, they help protect against more severe illness and death.
It’s important to note that scientists around the world are also closely monitoring Omicron’s severity While this variant appears to be highly transmissible, and it is still early for rigorous conclusions, the initial research indicates this variant may actually produce milder illness than Delta, which is currently the dominant strain in the United States.
But there’s still a tremendous amount of research to be done that could change how we view Omicron. This research will take time and patience.
What won’t change, though, is that vaccines are the best way to protect yourself and others against COVID-19. (And these recent data provide an even-stronger reason to get a booster now if you are eligible.) Wearing a mask, especially in public indoor settings, offers good protection against the spread of all SARS-CoV-2 variants. If you’ve got symptoms or think you may have been exposed, get tested and stay home if you get a positive result. As we await more answers, it’s as important as ever to use all the tools available to keep yourself, your loved ones, and your community happy and healthy this holiday season.
References:
[1] SARS-CoV-2 Omicron has extensive but incomplete escape of Pfizer BNT162b2 elicited neutralization and requires ACE2 for infection. Sandile C, et al. Sandile C, et al. medRxiv preprint. December 9, 2021.
[2] Pfizer and BioNTech provide update on Omicron variant. Pfizer. December 8, 2021.
[3] Effectiveness of COVID-19 vaccines against the Omicron (B.1.1.529) variant of concern. Andrews N, et al. KHub.net preprint. December 10, 2021.
Links:
COVID-19 Research (NIH)
Sigal Lab (Africa Health Research Institute, Durban, South Africa)
New Clues to Delta Variant’s Spread in Studies of Virus-Like Particles
Posted on by Dr. Francis Collins

About 70,000 people in the United States are diagnosed with COVID-19 each and every day. It’s clear that these new cases are being driven by the more-infectious Delta variant of SARS-CoV-2, the novel coronavirus that causes COVID-19. But why does the Delta variant spread more easily than other viral variants from one person to the next?
Now, an NIH-funded team has discovered at least part of Delta’s secret, and it’s not all attributable to those widely studied mutations in the spike protein that links up to human cells through the ACE2 receptor. It turns out that a specific mutation found within the N protein coding region of the Delta genome also enables the virus to pack more of its RNA code into the infected host cell. As a result, there is increased production of fully functional new viral particles, which can go on to infect someone else.
This finding, published in the journal Science [1], comes from the lab of Nobel laureate Jennifer Doudna at the Howard Hughes Medical Institute, the Gladstone Institutes, San Francisco, and the Innovative Genomics Institute at the University of California, Berkeley. Co-leading the team was Melanie Ott, Gladstone Institutes.
The Doudna and Ott teams have developed an exciting new tool to study variants of the coronavirus. It’s a lab construct called a virus-like particle (VLP). These specially made VLPs have all the structural proteins of SARS-CoV-2 (shown above), but they contain no genetic material. Consequently, they are non-infectious replicas of the real virus that can be studied safely in any lab. Scientists don’t have to reserve time in labs equipped with heightened levels of biosafety, as is required when working with whole virus.
The VLPs also allow researchers to explore changes found in the coronavirus’s other essential proteins, not just the spike protein on its surface. In fact, all of the SARS-CoV-2 variants of concern, as defined by the World Health Organization (WHO), carry at least one mutation within the same stretch of seven amino acids in a viral protein known as the nucleocapsid (N protein). This protein, which hasn’t been widely studied, is required for the virus to make more of itself. It is also involved in the virus’s ability to package and release infectious RNA.
In the Science paper, Doudna and colleagues took a closer look at the N protein. They did so by developing a special system that used VLPs to package and deliver viral RNA messages into human cells.
Here’s how it works: The VLPs include all four of SARS-CoV-2’s structural proteins, including the spike and N proteins. In addition, they contain the RNA sequence that allows the virus to recognize its genetic material within the cell, so that it can be packaged into the next generation of viral particles.
Though the particles look just like SARS-CoV-2 from the outside, they lack the vast majority of the viral genome on the inside. But they do have one other key component: a snippet of RNA that makes cells invaded by VLPs glow. In fact, the more RNA messages a VLP delivers, the brighter the cells will glow. It allowed the researchers to spot successful invasions, while also quantifying the amount of RNA a particular VLP packed into a cell.
The researchers then produced SARS-CoV-2 VLPs including four mutations that are universally found within the N proteins of more transmissible variants of concern. That’s when they discovered those variants produced and delivered 10 times more RNA messages into cells.
The increased RNA also fits with what has been observed in people infected with the Delta variant. They produce about 10 times more virus in their nose and throat compared to people infected with the older variants.
But did those findings match what happens in the real virus? To find out, the researchers and their colleagues tested the N protein mutation found in the Delta variant in a high-level biosafety lab. And, indeed, their studies showed that the mutated virus within infected human lung cells produced about 50 times more infectious virus compared to the original SARS-CoV-2 variant.
The findings suggest that the N protein could be an important new target for effective COVID-19 therapeutics, and that tracking newly emerging mutations in the N protein might also be important for identifying new viral variants of concern. This new system is a powerful tool, and one that can also be used for exploring how newly arising variants in the future might affect the course of this terrible pandemic.
Reference:
[1] Rapid assessment of SARS-CoV-2 evolved variants using virus-like particles. Syed AM, Taha TY, Tabata T, Chen IP, Ciling A, Khalid MM, Sreekumar B, Chen PY, Hayashi JM, Soczek KM, Ott M, Doudna JA. Science. 2021 Nov 4:eabl6184.
Links:
COVID-19 Research (NIH)
NIH Support: National Institute of Allergy and Infectious Diseases
A Real-World Look at COVID-19 Vaccines Versus New Variants
Posted on by Dr. Francis Collins

Clinical trials have shown the COVID-19 vaccines now being administered around the country are highly effective in protecting fully vaccinated individuals from the coronavirus SARS-CoV-2. But will they continue to offer sufficient protection as the frequency of more transmissible and, in some cases, deadly emerging variants rise?
More study and time is needed to fully answer this question. But new data from Israel offers an early look at how the Pfizer/BioNTech vaccine is holding up in the real world against coronavirus “variants of concern,” including the B.1.1.7 “U.K. variant” and the B.1.351 “South African variant.” And, while there is some evidence of breakthrough infections, the findings overall are encouraging.
Israel was an obvious place to look for answers to breakthrough infections. By last March, more than 80 percent of the country’s vaccine-eligible population had received at least one dose of the Pfizer/BioNTech vaccine. An earlier study in Israel showed that the vaccine offered 94 percent to 96 percent protection against infection across age groups, comparable to the results of clinical trials. But it didn’t dig into any important differences in infection rates with newly emerging variants, post-vaccination.
To dig a little deeper into this possibility, a team led by Adi Stern, Tel Aviv University, and Shay Ben-Shachar, Clalit Research Institute, Tel Aviv, looked for evidence of breakthrough infections in several hundred people who’d had at least one dose of the Pfizer/BioNTech vaccine [1]. The idea was, if this vaccine were less effective in protecting against new variants of concern, the proportion of infections caused by them should be higher in vaccinated compared to unvaccinated individuals.
During the study, reported as a pre-print in MedRxiv, it became clear that B.1.1.7 was the predominant SARS-CoV-2 variant in Israel, with its frequency increasing over time. By comparison, the B.1.351 “South African” variant was rare, accounting for less than 1 percent of cases sampled in the study. No other variants of concern, as defined by the World Health Organization, were detected.

In total, the researchers sequenced SARS-CoV-2 from more than 800 samples, including vaccinated individuals and matched unvaccinated individuals with similar characteristics including age, sex, and geographic location. They identified nearly 250 instances in which an individual became infected with SARS-CoV-2 after receiving their first vaccine dose, meaning that they were only partially protected. Almost 150 got infected sometime after receiving the second dose.
Interestingly, the evidence showed that these breakthrough infections with the B.1.1.7 variant occurred slightly more often in people after the first vaccine dose compared to unvaccinated people. No evidence was found for increased breakthrough rates of B.1.1.7 a week or more after the second dose. In contrast, after the second vaccine dose, infection with the B.1.351 became slightly more frequent. The findings show that people remain susceptible to B.1.1.7 following a single dose of vaccine. They also suggest that the two-dose vaccine may be slightly less effective against B.1.351 compared to the original or B.1.1.7 variants.
It’s important to note, however, that the researchers only observed 11 infections with the B.1.351 variant—eight of them in individuals vaccinated with two doses. Interestingly, all eight tested positive seven to 13 days after receiving their second dose. No one in the study tested positive for this variant two weeks or more after the second dose.
Many questions remain, including whether the vaccines reduced the duration and/or severity of infections. Nevertheless, the findings are a reminder that—while these vaccines offer remarkable protection—they are not foolproof. Breakthrough infections can and do occur.
In fact, in a recent report in the New England Journal of Medicine, NIH-supported researchers detailed the experiences of two fully vaccinated individuals in New York who tested positive for COVID-19 [2]. Though both recovered quickly at home, genomic data in those cases revealed multiple mutations in both viral samples, including a variant first identified in South Africa and Brazil, and another, which has been spreading in New York since November.
These findings in Israel and the United States also highlight the importance of tracking coronavirus variants and making sure that all eligible individuals get fully vaccinated as soon as they have the opportunity. They show that COVID-19 testing will continue to play an important role, even in those who’ve already been vaccinated. This is even more important now as new variants continue to rise in frequency.
Just over 100 million Americans aged 18 and older—about 40 percent of adults—are now fully vaccinated [3]. However, we need to get that number much higher. If you or a loved one haven’t yet been vaccinated, please consider doing so. It will help to save lives and bring this pandemic to an end.
References:
[1] Evidence for increased breakthrough rates of SARS-CoV-2 variants of concern in BNT162b2 mRNA vaccinated individuals. Kustin T et al. medRxiv. April 16, 2021.
[2] Vaccine breakthrough infections with SARS-CoV-2 variants. Hacisuleyman E, Hale C, Saito Y, Blachere NE, Bergh M, Conlon EG, Schaefer-Babajew DJ, DaSilva J, Muecksch F, Gaebler C, Lifton R, Nussenzweig MC, Hatziioannou T, Bieniasz PD, Darnell RB. N Engl J Med. 2021 Apr 21.
[3] COVID-19 vaccinations in the United States. Centers for Disease Control and Prevention.
Links:
COVID-19 Research (NIH)
Stern Lab (Tel Aviv University, Israel)
Ben-Shachar Lab (Clalit Research Institute, Tel Aviv, Israel)
NIH Support: National Institute of Allergy and Infectious Diseases
Finding New Ways to Fight Coronavirus … From Studying Bats
Posted on by Dr. Francis Collins

David Veesler has spent nearly 20 years imaging in near-atomic detail the parts of various viruses, including coronaviruses, that enable them to infect Homo sapiens. In fact, his lab at the University of Washington, Seattle, was the first to elucidate the 3D architecture of the now infamous spike protein, which coronaviruses use to gain entry into human cells [1]. He uses these fundamental insights to guide the design of vaccines and therapeutics, including promising monoclonal antibodies.
Now, Veesler and his lab are turning to another mammal in their search for new leads for the next generation of antiviral treatments, including ones aimed at the coronavirus that causes COVID-19, SARS-CoV-2. With support from a 2020 NIH Director’s Pioneer Award, Veesler will study members of the order Chiroptera. Or, more colloquially, bats.
Why bats? Veesler says bats are remarkable creatures. They are the only mammals capable of sustained flight. They rarely get cancer and live unusually long lives for such small creatures. More importantly for Veesler’s research, bats host a wide range of viruses—more than any other mammal species. Despite carrying all of these viruses, bats rarely show symptoms of being sick. Yet they are the source for many of the viruses that have spilled over into humans with devastating effect, including rabies, Ebola virus, Nipah and Hendra viruses, severe acute respiratory syndrome coronavirus (SARS-CoV), and, likely, SARS-CoV-2.
Beyond what is already known about bats’ intriguing qualities, Veesler says humans still have much to discover about these flying mammals, including how their immune systems cope with such an onslaught of viral invaders. For example, it turns out that a bat’s learned, or adaptive, immune system is, for the most part, uncharted territory. As such, it offers an untapped source of potentially promising viral inhibitors just waiting to be unearthed, fully characterized, and then used to guide the development of new kinds of anti-viral therapeutics.
In his studies, Veesler will work with collaborators studying bats around the world to characterize their antibody production. He wants to learn how these antibodies contribute to bats’ impressive ability to tolerate viruses and other pathogens. What is it about the structure of bat antibodies that make them different from human antibodies? And, how can those structural differences serve as blueprints for promising new treatments to combat many potentially deadly viruses?
Interestingly, Veesler’s original grant proposal makes no mention of SARS-CoV-2 or COVID-19. That’s because he submitted it just months before the first reports of the novel coronavirus in Wuhan, China. But Veesler doesn’t consider himself a visionary by expanding his research to bats. He and others had been working on closely related coronaviruses for years, inspired by earlier outbreaks, including SARS in 2002 and Middle East respiratory syndrome (MERS) in 2012 (although MERS apparently came from camels). The researcher didn’t see SARS-CoV-2 coming, but he recognized the potential for some kind of novel coronavirus outbreak in the future.
These days, the Veesler lab has been hard at work to understand SARS-CoV-2 and the human immune response to the virus. His team showed that SARS-CoV-2 uses the human receptor ACE2 to gain entry into our cells [2]. He’s also a member of the international research team that identified a human antibody, called S309, from a person who’d been infected with SARS in 2003. This antibody is showing promise for treating COVID-19 [3], now in a phase 3 clinical trial in the United States.
In another recent study, reported as a pre-print in bioRxiv, Veesler’s team mapped dozens of distinct human antibodies capable of neutralizing SARS-CoV-2 by their ability to hit viral targets outside of the well-known spike protein [4]. Such discoveries may form the basis for new and promising combinations of antibodies to treat COVID-19 that won’t be disabled by concerning new variations in the SARS-CoV-2 spike protein. Perhaps, in the future, such therapeutic cocktails may include modified bat-inspired antibodies too.
References:
[1] Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Walls AC, Tortorici MA, Bosch BJ, Frenz B, Rottier PJM, DiMaio F, Rey FA, Veesler D. Nature. 2016 Mar 3;531(7592):114-117.
[2] Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Cell. 2020 Apr 16;181(2):281-292.e6.
[3] Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Pinto D, Park YJ, Beltramello M, Veesler D, Cortil D, et al. Nature.18 May 2020 [Epub ahead of print]
[4] N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. McCallum M, Marco A, Lempp F, Tortorici MA, Pinto D, Walls AC, Whelan SPJ, Virgin HW, Corti D, Pizzuto MS, Veesler D, et al. bioRxiv. 2021 Jan 14.
Links:
COVID-19 Research (NIH)
Veesler Lab (University of Washington, Seattle)
Veesler Project Information (NIH RePORTER)
NIH Director’s Pioneer Award Program (Common Fund)
NIH Support: Common Fund; National Institute of Allergy and Infectious Diseases
South Africa Study Shows Power of Genomic Surveillance Amid COVID-19 Pandemic
Posted on by Dr. Francis Collins
Considerable research is underway around the world to monitor the spread of new variants of SARS-CoV-2, the coronavirus that causes COVID-19. That includes the variant B.1.351 (also known as 501Y.V2), which emerged in South Africa towards the end of 2020 [1, 2]. Public health officials in South Africa have been busy tracing the spread of this genomic variant and others across their country. And a new analysis of such data reveals that dozens of distinct coronavirus variants were already circulating in South Africa well before the appearance of B.1.351.
A study of more than 1,300 near-whole genome sequences of SARS-CoV-2, published recently in the journal Nature Medicine, shows there were in fact at least 42 SARS-CoV-2 variants spreading in South Africa within the pandemic’s first six months in that country [3]. Among them were 16 variants that had never before been described. Most of the single-letter changes carried by these variants didn’t change the virus in important ways and didn’t rise to significant frequency. But the findings come as another critical reminder of the value of genomic surveillance to track the spread of SARS-CoV-2 to identify any potentially worrisome new variants and to inform measures to get this devastating pandemic under control.
SARS-CoV-2 was first detected in South Africa on March 5, 2020, in a traveler returning from Italy. By November 2020, despite considerable efforts to slow the spread, more than 785,000 people in South Africa were infected, accounting for about half of all reported COVID-19 cases on the African continent.
Recognizing the importance of genomic surveillance, researchers led by Houriiyah Tegally and Tulio de Oliveira, University of KwaZulu-Natal, Durban, South Africa, wasted no time in producing 1,365 near-complete SARS-CoV-2 genomes by mid-September, near the end of the coronavirus’s first peak in the country. Those samples had been collected in hundreds of clinics over the course of the pandemic in eight of South Africa’s nine provinces, offering a broad picture of the spread and emergence of new variants across the country.
The data revealed three main variants, dubbed B.1.1.54, B.1.1.56, and C.1, that were responsible for 42 percent of all the infections in South Africa’s first wave. Of the 16 newly described variants, most carried single-letter changes that haven’t been identified in other countries.
The majority of changes were what scientists refer to as “synonymous,” meaning that they don’t change the structure or function of any of the virus’s essential proteins. The exception is the newly identified C.1, which includes 16 single-letter changes compared to the original sequence from Wuhan, China. One of those 16 changes swaps a single amino acid for another on SARS-CoV-2’s spike protein. That’s notable because the spike protein is a key target of antibodies and also is essential to the virus’s ability to infect human cells.
In fact, four of the most prevalent variants in South Africa all carry this same mutation. The researchers also saw three other changes that would alter the spike protein in different ways, although the significance of these for viral spread and our efforts to stop it isn’t yet clear.
Importantly, the data show that the bulk of introductions to South Africa happened early on, before lockdown and travel restrictions were implemented in late March. Subsequently, much of the spread within South Africa stemmed from hospital outbreaks. For example, an outbreak of the C.1 variant in the North West Province in April ultimately led this variant to become the most geographically widespread in South Africa by the end of August. Meanwhile, an earlier identified South African-specific variant, B.1.106, first identified in April, vanished altogether after outbreaks were controlled in KwaZulu-Natal Province, where the researchers reside.
Genomic surveillance has remarkable power for understanding the evolution of SARS-CoV-2 and tracking the dynamics of its transmission. Tegally and de Oliveira’s team notes that this type of intensive genomic surveillance now can be used on a large scale across Africa and around the world to identify new variants of SARS-CoV-2 and to develop timely measures to control the spread of the virus. They’re now working with the African CDC to expand genomic surveillance across Africa [4].
Such genomic surveillance was crucial in the subsequent identification of the B.1.351 variant in South Africa that we’ve been hearing so much about, with its potential to evade our current treatments and vaccines. By picking up on such concerning mutations early through genomic surveillance and understanding how the virus is spreading over time and space, the hope is we’ll be better informed and more adept in our efforts to get this pandemic under control.
References:
[1] Emerging SARS-CoV-2 variants. Centers for Disease Control and Prevention.
[2] Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Bhiman J, Williamson C, de Oliveira T, et al. medRxiv 2020 Dec 22.
[3] Sixteen novel lineages of SARS-CoV-2 in South Africa. Tegally H, Wilkinson E, Lessells RJ, Giandhari J, Pillay S, Msomi N, Mlisana K, Bhiman JN, von Gottberg A, Walaza S, Fonseca V, Allam M, Ismail A, Glass AJ, Engelbrecht S, Van Zyl G, Preiser W, Williamson C, Petruccione F, Sigal A, Gazy I, Hardie D, Hsiao NY, Martin D, York D, Goedhals D, San EJ, Giovanetti M, Lourenço J, Alcantara LCJ, de Oliveira T. Nat Med. 2021 Feb 2.
[4] Accelerating genomics-based surveillance for COVID-19 response in Africa. Tessema SK, Inzaule SC, Christoffels A, Kebede Y, de Oliveira T, Ouma AEO, Happi CT, Nkengasong JN.Lancet Microbe. 2020 Aug 18.
Links:
COVID-19 Research (NIH)
Houriiyah Tegally (University of KwaZulu-Natal, Durban, South Africa)
Tulio de Oliveira (University of KwaZulu-Natal)
Creative Minds: Interpreting Your Genome
Posted on by Dr. Francis Collins

Credit: Jane Ades, National Human Genome Research Institute, NIH
Just this year, we’ve reached the point where we can sequence an entire human genome for less than $1,000. That’s great news—and rather astounding, since the first human genome sequence (finished in 2003) cost an estimated $400,000,000! Does that mean we’ll be able to use each person’s unique genetic blueprint to guide his or her health care from cradle to grave? Maybe eventually, but it’s not quite as simple as it sounds.
Before we can use your genome to develop more personalized strategies for detecting, treating, and preventing disease, we need to be able to interpret the many variations that make your genome distinct from everybody else’s. While most of these variations are neither bad nor good, some raise the risk of particular diseases, and others serve to lower the risk. How do we figure out which is which?
Jay Shendure, an associate professor at the University of Washington in Seattle, has an audacious plan to figure this out, which is why he is among the 2013 recipients of the NIH Director’s Pioneer Award.