protein
Artificial Intelligence Accurately Predicts Protein Folding
Posted on by Dr. Francis Collins
Proteins are the workhorses of the cell. Mapping the precise shapes of the most important of these workhorses helps to unlock their life-supporting functions or, in the case of disease, potential for dysfunction. While the amino acid sequence of a protein provides the basis for its 3D structure, deducing the atom-by-atom map from principles of quantum mechanics has been beyond the ability of computer programs—until now.
In a recent study in the journal Science, researchers reported they have developed artificial intelligence approaches for predicting the three-dimensional structure of proteins in record time, based solely on their one-dimensional amino acid sequences [1]. This groundbreaking approach will not only aid researchers in the lab, but guide drug developers in coming up with safer and more effective ways to treat and prevent disease.
This new NIH-supported advance is now freely available to scientists around the world. In fact, it has already helped to solve especially challenging protein structures in cases where experimental data were lacking and other modeling methods hadn’t been enough to get a final answer. It also can now provide key structural information about proteins for which more time-consuming and costly imaging data are not yet available.
The new work comes from a group led by David Baker and Minkyung Baek, University of Washington, Seattle, Institute for Protein Design. Over the course of the pandemic, Baker’s team has been working hard to design promising COVID-19 therapeutics. They’ve also been working to design proteins that might offer promising new ways to treat cancer and other conditions. As part of this effort, they’ve developed new computational approaches for determining precisely how a chain of amino acids, which are the building blocks of proteins, will fold up in space to form a finished protein.
But the ability to predict a protein’s precise structure or shape from its sequence alone had proven to be a difficult problem to solve despite decades of effort. In search of a solution, research teams from around the world have come together every two years since 1994 at the Critical Assessment of Structure Prediction (CASP) meetings. At these gatherings, teams compete against each other with the goal of developing computational methods and software capable of predicting any of nature’s 200 million or more protein structures from sequences alone with the greatest accuracy.
Last year, a London-based company called DeepMind shook up the structural biology world with their entry into CASP called AlphaFold. (AlphaFold was one of Science’s 2020 Breakthroughs of the Year.) They showed that their artificial intelligence approach—which took advantage of the 170,000 proteins with known structures in a reiterative process called deep learning—could predict protein structure with amazing accuracy. In fact, it could predict most protein structures almost as accurately as other high-resolution protein mapping techniques, including today’s go-to strategies of X-ray crystallography and cryo-EM.
The DeepMind performance showed what was possible, but because the advances were made by a world-leading deep learning company, the details on how it worked weren’t made publicly available at the time. The findings left Baker, Baek, and others eager to learn more and to see if they could replicate the impressive predictive ability of AlphaFold outside of such a well-resourced company.
In the new work, Baker and Baek’s team has made stunning progress—using only a fraction of the computational processing power and time required by AlphaFold. The new software, called RoseTTAFold, also relies on a deep learning approach. In deep learning, computers look for patterns in large collections of data. As they begin to recognize complex relationships, some connections in the network are strengthened while others are weakened. The finished network is typically composed of multiple information-processing layers, which operate on the data to return a result—in this case, a protein structure.
Given the complexity of the problem, instead of using a single neural network, RoseTTAFold relies on three. The three-track neural network integrates and simultaneously processes one-dimensional protein sequence information, two-dimensional information about the distance between amino acids, and three-dimensional atomic structure all at once. Information from these separate tracks flows back and forth to generate accurate models of proteins rapidly from sequence information alone, including structures in complex with other proteins.
As soon as the researchers had what they thought was a reasonable working approach to solve protein structures, they began sharing it with their structural biologist colleagues. In many cases, it became immediately clear that RoseTTAFold worked remarkably well. What’s more, it has been put to work to solve challenging structural biology problems that had vexed scientists for many years with earlier methods.
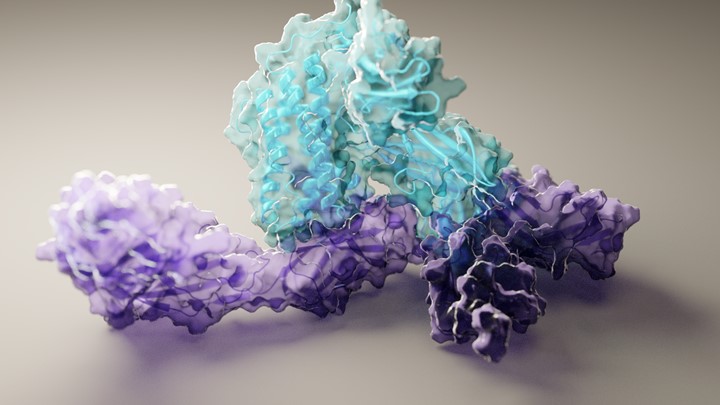
RoseTTAFold already has solved hundreds of new protein structures, many of which represent poorly understood human proteins. The 3D rendering of a complex showing a human protein called interleukin-12 in complex with its receptor (above image) is just one example. The researchers have generated other structures directly relevant to human health, including some that are related to lipid metabolism, inflammatory conditions, and cancer. The program is now available on the web and has been downloaded by dozens of research teams around the world.
Cryo-EM and other experimental mapping methods will remain essential to solve protein structures in the lab. But with the artificial intelligence advances demonstrated by RoseTTAFold and AlphaFold, which has now also been released in an open-source version and reported in the journal Nature [2], researchers now can make the critical protein structure predictions at their desktops. This newfound ability will be a boon to basic science studies and has great potential to speed life-saving therapeutic advances.
References:
[1] Accurate prediction of protein structures and interactions using a three-track neural network. Baek M, DiMaio F, Anishchenko I, Dauparas J, Grishin NV, Adams PD, Read RJ, Baker D., et al. Science. 2021 Jul 15:eabj8754.
[2] Highly accurate protein structure prediction with AlphaFold. Jumper J, Evans R, Pritzel A, Green T, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. et al. Nature. 2021 Jul 15.
Links:
Structural Biology (National Institute of General Medical Sciences/NIH)
The Structures of Life (NIGMS)
Baker Lab (University of Washington, Seattle)
CASP 14 (University of California, Davis)
NIH Support: National Institute of Allergy and Infectious Diseases; National Institute of General Medical Sciences
Adding Letters to the DNA Alphabet
Posted on by Dr. Francis Collins
Credit: William B. Kiosses
The recipes for life, going back billions of years to the earliest single-celled organisms, are encoded in a DNA alphabet of just four letters. But is four as high as the DNA code can go? Or, as researchers have long wondered, is it chemically and biologically possible to expand the DNA code by a couple of letters?
A team of NIH-funded researchers is now answering these provocative questions. The researchers recently engineered a semi-synthetic bacterium containing DNA with six letters, including two extra nucleotides [1, 2]. Now, in a report published in Nature, they’ve taken the next critical step [3]. They show that bacteria, like those in the photo, are not only capable of reliably passing on to the next generation a DNA code of six letters, they can use that expanded genetic information to produce novel proteins unlike any found in nature.
Creative Minds: Interpreting Your Genome
Posted on by Dr. Francis Collins

Credit: Jane Ades, National Human Genome Research Institute, NIH
Just this year, we’ve reached the point where we can sequence an entire human genome for less than $1,000. That’s great news—and rather astounding, since the first human genome sequence (finished in 2003) cost an estimated $400,000,000! Does that mean we’ll be able to use each person’s unique genetic blueprint to guide his or her health care from cradle to grave? Maybe eventually, but it’s not quite as simple as it sounds.
Before we can use your genome to develop more personalized strategies for detecting, treating, and preventing disease, we need to be able to interpret the many variations that make your genome distinct from everybody else’s. While most of these variations are neither bad nor good, some raise the risk of particular diseases, and others serve to lower the risk. How do we figure out which is which?
Jay Shendure, an associate professor at the University of Washington in Seattle, has an audacious plan to figure this out, which is why he is among the 2013 recipients of the NIH Director’s Pioneer Award.
MicroRNA Research Takes Aim at Cholesterol
Posted on by Dr. Francis Collins
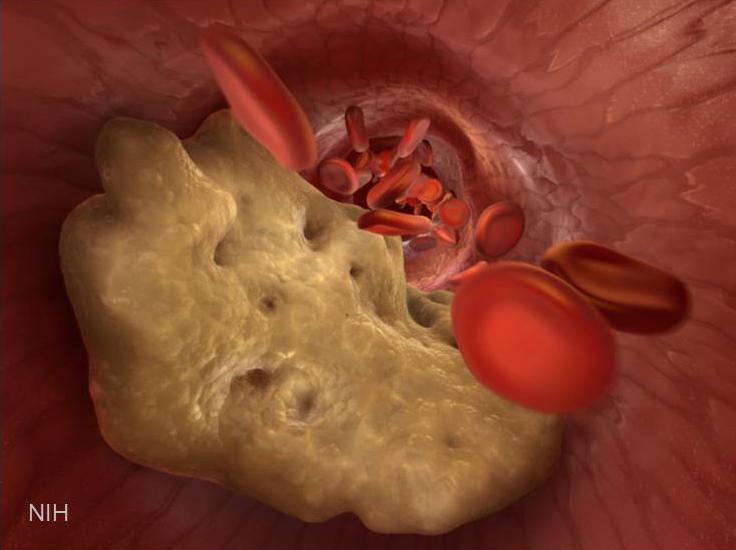
Caption: Illustration of artery partially blocked by a cholesterol plaque.
If you’re concerned about your cardiovascular health, you’re probably familiar with “good” and “bad” cholesterol: high-density lipoprotein (HDL) and its evil counterpart, low-density lipoprotein (LDL). Too much LDL floating around in your blood causes problems by sticking to the artery walls, narrowing the passage and raising risk of a stroke or heart attack. Statins work to lower LDL. HDL, on the other hand, cruises through your arteries scavenging excess cholesterol and returning it to the liver, where it’s broken down.
Gain Without Pain: New Clues for Analgesic Design
Posted on by Dr. Francis Collins

Photo Credit: Matthew Rowe, Michigan State University
If you’re a southern grasshopper mouse, nothing beats a delicious snack of scorpion. But what, you might ask, prevents that from being a painful or even fatal event? Well, this native of the Arizona desert has evolved an amazing resistance to the stings of the bark scorpion—stings so painful and toxic they kill house mice and other rodents of similar size.
Why am I sharing this bit of natural history? Well, it turns out that by studying the grasshopper mouse and its unusual diet, NIH-funded researchers at the Indiana University School of Medicine and collaborators at the University of Texas, Austin, have identified a new target on nerve fibers that could lead to more effective and less addictive pain medications for humans.
Yeast Reveals New Drug Target for Parkinson’s
Posted on by Dr. Francis Collins

Credit: Daniel Tardiff, Whitehead Institute
Many progressive neurodegenerative disorders like Alzheimer’s, Huntington’s, and Parkinson’s disease, are characterized by abnormal clumps of proteins that clog up the cell and disrupt normal cellular functions. But it’s difficult to study these complex disease processes directly in the brain—so NIH-funded researchers, led by a team at the Whitehead Institute for Biomedical Research, Cambridge, MA, have turned to yeast for help.
Now, it may sound odd to study a brain disease in yeast, a microorganism long used in baking and brewing. After all, the brain is made up of billions of cells of many different types, while yeast grows as a single cell. But because the processes of protein production are generally conserved from yeast to humans, we can use this infinitely simpler organism to figure out what the proteins clumps are doing and test various drug candidates to halt the damage.
Less TOR Protein Extends Mouse Lifespan
Posted on by Dr. Francis Collins
 The average life expectancy in the United States currently is about 79 [1]. And, unsurprisingly, more than two-thirds of Americans say they’d like to live another 10 to 20 years longer [2].
The average life expectancy in the United States currently is about 79 [1]. And, unsurprisingly, more than two-thirds of Americans say they’d like to live another 10 to 20 years longer [2].
One possible route to a longer life is to cut calories drastically. Not much fun perhaps, but there’s evidence it works in yeast, worms, and mice—but probably not in monkeys [3]. The potential life-extending strategy that I’d like to tell you about today focuses on the drug rapamycin, which blocks the activity of a protein called “target of rapamycin,” or TOR. Recently, a team here at NIH discovered that—at least in mice—reducing production of this protein through genetic engineering can add about 20% to the lifespan [4].
Protein Pile-up: Common Cause of Brain Disease
Posted on by Dr. Francis Collins

Caption: Left: High levels of the toxic ataxin-1 protein have destroyed nerve cells in the cerebellum of a mouse, causing a severe disease. Right: Here researchers have genetically blocked the genes that normally produce high levels of ataxin-1. This prevents the disease from developing and keeps the brain healthy.
Credit: Harry Orr, Department of Laboratory Medicine and Pathology, University of Minnesota
With our aging population, more people are developing neurodegenerative disorders like Alzheimer’s and Parkinson’s disease. We currently don’t know how to prevent or cure these conditions, and their increasing prevalence not only represents a tragedy for affected individuals and their families, but also a looming public health and economic crisis.
Even though neurodegenerative diseases have varied roots—and affect distinct cell types in different brain regions—they do share something in common. In most of these disorders, we see some type of toxic protein accumulating in the brain. It’s as if the brain’s garbage disposal system is blocked, letting the waste pile up. In Huntington’s disease, huntingtin is the disease-causing protein. In spinocerebellar ataxia, it’s the ataxins. In Alzheimer’s, it’s beta-amyloid; in Parkinson’s, it’s α-synuclein. When garbage builds up in your kitchen, it’s a bad situation. When it’s in your brain, the consequences are deadly.
Last week, a team of NIH-funded researchers based at the Baylor College of Medicine in Texas and at the University of Minnesota revealed a clever way to identify genes that normally increase the levels of these rogue disease-causing proteins.
Brown Fat, White Fat, Good Fat, Bad Fat
Posted on by Dr. Francis Collins

Credit: Patrick Seale, University of Pennsylvania School of Medicine
Fat has been villainized; but all fat was not created equal. Our two main types of fat—brown and white—play different roles. Now, two teams of NIH-funded researchers have enriched our understanding of adipose tissue. The first team discovered the genetic switch that triggers the development of brown fat [1], and the second figured out how the body can recruit white fat and transform it into brown [2].
Why would we want to change white fat into brown? White fat stores energy as large fat droplets, while brown fat has much smaller droplets and is specialized to burn them, yielding heat. Brown fat cells are packed with energy generating powerhouses called mitochondria that contain iron—which gives them their brown color. Infants are born with rich stores of brown fat (about 5% of total body mass) on the upper spine and shoulders to keep them warm. It used to be thought that brown fat disappeared by adulthood—but it turns out we harbor small reserves in our shoulders and neck.
New Insight into Parkinson’s Disease
Posted on by Dr. Francis Collins

Credit: Samantha Orenstein and Dr. Esperanza Arias, Department of Developmental and Molecular Biology, Albert Einstein College of Medicine, Bronx, New York
I’m blogging today to tell you about a new NIH funded report [1] describing a possible cause of Parkinson’s disease: a clog in the protein disposal system.
You probably already know something about Parkinson’s disease. Many of us know individuals who have been stricken, and actor Michael J. Fox, who suffers from it, has done a great job talking about and spreading awareness of it. Parkinson’s is a progressive neurodegenerative condition in which the dopamine-producing cells in the brain region called the substantia nigra begin to sicken and die. These cells are critical for controlling movement; their death causes shaking, difficulty moving, and the characteristic slow gait. Patients can have trouble swallowing, chewing, and speaking. As the disease progresses, cognitive and behavioral problems take hold—depression, personality shifts, sleep disturbances.
Next Page