travel
Planning Your Holidays During the COVID-19 Pandemic
Posted on by Dr. Francis Collins

With the holiday season fast approaching and coronavirus disease 2019 (COVID-19) surging in most parts of the country, millions of Americans—including me and my family—will break with tradition this year to celebrate in ways that we hope will help to keep us all safe and healthy. Granted, this may present some difficult emotional and logistical challenges, but I’m confident that the American can-do spirit will rise to meet those challenges.
I also recognize that this will be hard for many of us. Celebrating holidays alone or with your immediate household members can sound rather dreary. After all, who wants to roast and carve a turkey for just a few people? But, if you look at it another way, the pandemic does offer opportunities to make this holiday a season to remember in new and different ways. Here are a couple of ideas that you may want to consider:
Send Gifts. Although COVID-19 has changed our lives in many ways, sending cards or gifts remains a relatively easy way to let loved ones know that you’re thinking of them. Who wouldn’t want to receive some home-baked goodies, a basket of fresh fruit, or a festive wreath? If you enjoy knitting, candle making, or other ways of crafting gifts for the holidays, now’s the time to start planning for Thanksgiving through the New Year.
Make Videos. When I’m visiting family, there is often music involved—with guitar, piano, and maybe some singing. But, this year, I’ll have to be content with video recording a few songs and sending them to others by text or email. Come to think of it, the kids and the grandkids might enjoy these songs just as much—or even more—if they can watch them at a time and place that works best for them. (On the other hand, some of them might roll their eyes and decide not to open that video file!) If you don’t play a guitar or like to sing, you can still make your own holiday-themed videos. Maybe share a dance routine, a demonstration of athletic skill, or even some stand-up comedy. The key is to have fun and let your imagination run free.
Share a Meal Remotely. Most of our end-of-the-year holidays involve the family sitting around a table overflowing with delicious food. With all of the videoconferencing platforms now available, it is easy to set aside a block of time to share a meal and good conversation remotely with friends and family members, whether they live nearby or across the country. Rather than one cook slaving over a hot stove or a certain person monopolizing the dinner table conversation, everyone gets a chance to cook and share their stories via their smartphone, tablet, or laptop. You can compare your culinary creations, swap recipes, and try to remember to leave room for dessert. If you have a tradition of playing games or giving thanks for your many blessings, you can still do many of these activities remotely.
Take an After-Dinner Walk. Due to the physical demands and psychological impacts of the COVID-19 pandemic, it’s been difficult for many of us to stay physically active. The key is making exercise a daily priority, and the holidays are no different. After your holiday meal, go on a virtual group walk through your respective neighborhoods to work off the food. Thanks to your smartphone’s camera, you can share your time outdoors and all of the interesting sights along the way. (Yes, the new playground in the local park looks fantastic, and the neighbors really did just paint their house purple!)
Stay Safe. If you plan to go ahead and join a holiday gathering in person, it’s important to remain vigilant, even when interacting with dear friends and loved ones. The greatest risk for spread of COVID-19 right now is these family gatherings. Remember there are risks associated with travel and with interacting with people who’ve not been tested for the coronavirus prior to the event, especially if they reside in a COVID hot spot—which is almost everywhere these days. Try to keep any family gatherings brief and relatively small, about five people or less. If the weather permits, hold the get-together outdoors.
To protect yourself and your loved ones, both now and over the holidays, please follow these 3 W’s:
• Wear a mask when you are out in public and when you are indoors with people who are not part of your immediate household. The only exception is while eating or drinking!
• Watch your distance, staying at least 6 feet away from people who are not part of your immediate household.
• Wash your hands thoroughly and frequently.
Making all of these adjustments is a lot to consider when you’re trying to have a good time and there are children and older adults in the mix. That’s why I and my wife Diane decided the best plan for us this holiday season is to stay home in Maryland and forgo our traditional trips to family in Michigan and North Carolina. Not only did we want to reduce the risk of possibly contracting COVID-19 from—or transmitting it to—our faraway loved ones, we want to do everything we can to protect our local friends and co-workers from the coronavirus.
While this holiday season is likely to be memorable in ways that we never could have imagined, I’m confident that, thanks to the rapid advances being made by medical research, we ultimately will get the COVID-19 pandemic under control so we can once again give everyone we love a big hug in person. Until then, please stay safe. Wishing each of you a wonderful and healthful holiday season, starting with a Happy Thanksgiving!
Links:
Coronavirus (COVID) (NIH)
Your Health: Holiday Celebrations and Small Gatherings (Centers for Disease Control and Prevention, Atlanta)
Your Health: Personal and Social Activities (CDC)
Genome Data Help Track Community Spread of COVID-19
Posted on by Dr. Francis Collins
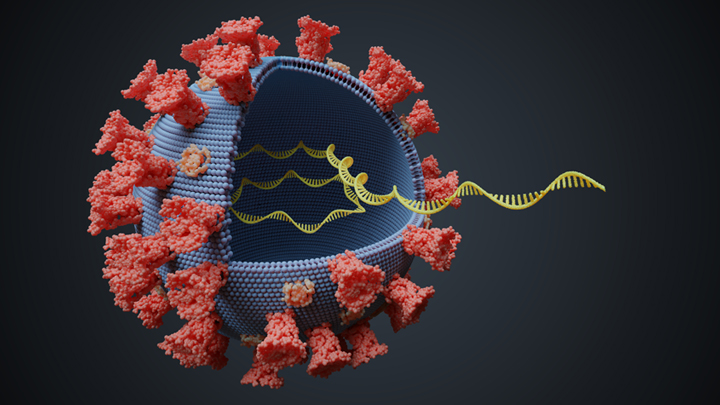
Contact tracing, a term that’s been in the news lately, is a crucial tool for controlling the spread of SARS-CoV-2, the novel coronavirus that causes COVID-19. It depends on quick, efficient identification of an infected individual, followed by identification of all who’ve recently been in close contact with that person so the contacts can self-quarantine to break the chain of transmission.
Properly carried out, contact tracing can be extremely effective. It can also be extremely challenging when battling a stealth virus like SARS-CoV-2, especially when the virus is spreading rapidly.
But there are some innovative ways to enhance contact tracing. In a new study, published in the journal Nature Medicine, researchers in Australia demonstrate one of them: assembling genomic data about the virus to assist contact tracing efforts. This so-called genomic surveillance builds on the idea that when the virus is passed from person to person over a few months, it can acquire random variations in the sequence of its genetic material. These unique variations serve as distinctive genomic “fingerprints.”
When COVID-19 starts circulating in a community, researchers can fingerprint the genomes of SARS-CoV-2 obtained from newly infected people. This timely information helps to tell whether that particular virus has been spreading locally for a while or has just arrived from another part of the world. It can also show where the viral subtype has been spreading through a community or, best of all, when it has stopped circulating.
The recent study was led by Vitali Sintchenko at the University of Sydney. His team worked in parallel with contact tracers at the Ministry of Health in New South Wales (NSW), Australia’s most populous state, to contain the initial SARS-CoV-2 outbreak from late January through March 2020.
The team performed genomic surveillance, using sequencing data obtained within about five days, to understand local transmission patterns. They also wanted to compare what they learned from genomic surveillance to predictions made by a sophisticated computer model of how the virus might spread amongst Australia’s approximately 24 million citizens.
Of the 1,617 known cases in Sydney over the three-month study period, researchers sequenced viral genomes from 209 (13 percent) of them. By comparing those sequences to others circulating overseas, they found a lot of sequence diversity, indicating that the novel coronavirus had been introduced to Sydney many times from many places all over the world.
They then used the sequencing data to better understand how the virus was spreading through the local community. Their analysis found that the 209 cases under study included 27 distinct genomic fingerprints. Based on the close similarity of their genomic fingerprints, a significant share of the COVID-19 cases appeared to have stemmed from the direct spread of the virus among people in specific places or facilities.
What was most striking was that the genomic evidence helped to provide information that contact tracers otherwise would have lacked. For instance, the genomic data allowed the researchers to identify previously unsuspected links between certain cases of COVID-19. It also helped to confirm other links that were otherwise unclear.
All told, researchers used the genomic evidence to cluster almost 40 percent of COVID-19 cases (81 of 209) for which the community-based data alone couldn’t identify a known contact source for the infection. That included 26 cases in which an individual who’d recently arrived in Australia from overseas spread the infection to others who hadn’t traveled. The genomic information also helped to identify likely sources in the community for another 15 locally acquired cases that weren’t known based on community data.
The researchers compared their genome surveillance data to SARS-CoV-2’s expected spread as modeled in a computer simulation based on travel to and from Australia over the time period in question. Because the study involved just 13 percent of all known COVID-19 cases in Sydney between late January through March, it’s not surprising that the genomic data presents an incomplete picture, detecting only a portion of the possible chains of transmission expected in the simulation model.
Nevertheless, the findings demonstrate the value of genomic data for tracking the virus and pinpointing exactly where in the community it is spreading. This can help to fill in important gaps in the community-based data that contact tracers often use. Even more exciting, by combining traditional contact tracing, genomic surveillance, and mathematical modeling with other emerging tools at our disposal, it may be possible to get a clearer picture of the movement of SARS-CoV-2 and put more targeted public health measures in place to slow and eventually stop its deadly spread.
Reference:
[1] Revealing COVID-19 transmission in Australia by SARS-CoV-2 genome sequencing and agent-based modeling. Rockett RJ, Arnott A, Lam C, et al. Nat Med. 2020 July 9. [Published online ahead of print]
Links:
Coronavirus (COVID-19) (NIH)
Vitali Sintchenko (University of Sydney, Australia)
Uncovering a Hidden Zika Outbreak in Cuba
Posted on by Dr. Francis Collins

When Brazilian health officials discovered four years ago that the mosquito-borne Zika virus could cause severe birth defects and other serious health problems, it prompted a major effort across the Americas to curb the infection by controlling mosquitoes and issuing travel advisories. By mid-2017, the hard work seemed to have paid off, and reports of new Zika infections had nearly stopped.
But it turns out Zika may be tougher to control than once thought. New research shows that a large, previously hidden outbreak of Zika virus disease occurred in Cuba, just when it looked like the worst of the epidemic was over. The finding suggests that the Zika virus can linger over long periods, and that mosquito control efforts alone may slow, but not necessarily stop, the march of this potentially devastating infectious disease.
When combating global epidemics, it’s critical to track the spread of dangerous viruses from one place to the next. But some viruses can be tougher to monitor than others, and that certainly has been the case with Zika in the Americas. Though the virus can harm unborn children, many people infected with Zika never feel lousy enough to go to the doctor. Those who do often have symptoms that overlap with other prevalent tropical diseases, such as dengue and chikungunya fever, making it hard to recognize Zika.
That’s why in Brazil, where Zika arrived in the Americas by early 2014, this unexpected viral intruder went undetected for well over a year. By then, it had spread unnoticed to Honduras, circulating rapidly to other Central American nations and Mexico—likely by late 2014 and into 2015.
In the United States, even with close monitoring, a small local outbreak of Zika virus in Florida also went undetected for about three months in 2016 [1]. Then, in 2017, Florida officials began noticing something strange: new cases of Zika infection in people who had traveled to Cuba.
This came as a real surprise because Cuba, unlike most other Caribbean islands, was thought to have avoided an outbreak. What’s more, by then the Zika epidemic in the Americas had slowed to a trickle, prompting the World Health Organization to delist it as a global public health emergency of international concern.
Given the Cuban observation, some wondered whether the Zika epidemic in the Americas was really over. Among them was an NIH-supported research team, including Nathan Grubaugh, Yale School of Public Health, New Haven, CT; Sharon Isern and Scott Michael, Florida Gulf Coast University, Fort Myers; and Kristian Andersen, The Scripps Research Institute, La Jolla, CA, who worked closely with the Florida Department of Health, including Andrea Morrison.
As published in Cell, the team was able to document a previously unreported outbreak in Cuba after the epidemic had seemingly ended [2]. Interestingly, another research group in Spain also recently made a similar observation about Zika in Cuba [3].
In the Cell paper, the researchers show that between June 2017 and October 2018, all but two of 155 cases—a whopping 98 percent of travel-associated Zika infections—traced back to Cuba. Further analysis suggests that the outbreak in Cuba was likely of similar magnitude to outbreaks that occurred in other Caribbean nations.
Their estimates suggest there were likely many thousands of Zika cases in Cuba, and more than 5,000 likely should have been diagnosed and reported in 2017. The only difference was the timing. The Cuban outbreak of Zika virus occurred about a year after infections subsided elsewhere in the Caribbean.
To fill in more of the blanks, the researchers relied on Zika virus genomes from nine infected Florida travelers who returned from Cuba in 2017 and 2018. The sequencing data support multiple introductions of Zika virus to Cuba from other Caribbean islands in the summer of 2016.
The outbreak peaked about a year after the virus made its way to Cuba, similar to what happened in other places. But the Cuban outbreak was likely delayed by a year thanks to an effective mosquito control campaign by local authorities, following detection of the Brazilian outbreak. While information is lacking, including whether Zika infections had caused birth defects, it’s likely those efforts were relaxed once the emergency appeared to be over elsewhere in the Caribbean, and the virus took hold.
The findings serve as yet another reminder that the Zika virus—first identified in the Zika Forest in Uganda in 1947 and for many years considered a mostly inconsequential virus [4]—has by no means been eliminated. Indeed, such unrecognized and delayed outbreaks of Zika raise the risk of travelers innocently spreading the virus to other parts of the world.
The encouraging news is that, with travel surveillance data and genomic tools —enabled by open science—it is now possible to detect such outbreaks. By combining resources and data, it will be possible to develop even more effective and responsive surveillance frameworks to pick up on emerging health threats in the future.
In the meantime, work continues to develop a vaccine for the Zika virus, with more than a dozen clinical trials underway that pursue a variety of vaccination strategies. With the Zika pandemic resolved in the Americas, these studies can be harder to conduct, since proof of efficacy is not possible without active infections. But, as this paper shows, we must remain ready for future outbreaks of this unique and formidable virus.
References:
[1] Genomic epidemiology reveals multiple introductions of Zika virus into the United States. Grubaugh et al. Nature. 2017 Jun 15;546(7658):401-405.
[2] Travel surveillance and genomics uncover a hidden Zika outbreak during the waning epidemic. Grubaugh ND, Saraf S, Gangavarapu K, Watts A, Tan AL, Oidtman RJ, Magnani DM, Watkins DI, Palacios G, Hamer DH; GeoSentinel Surveillance Network, Gardner LM, Perkins TA, Baele G, Khan K, Morrison A, Isern S, Michael SF, Andersen .KG, et. al. Cell. 2019 Aug 22;178(5):1057-1071.e11.
[3] Mirroring the Zika epidemics in Cuba: The view from a European imported diseases clinic. Almuedo-Riera A, Rodriguez-Valero N, Camprubí D, Losada Galván I, Zamora-Martinez C, Pousibet-Puerto J, Subirà C, Martinez MJ, Pinazo MJ, Muñoz J. Travel Med Infect Dis. 2019 Jul – Aug;30:125-127.
[4] Pandemic Zika: A Formidable Challenge to Medicine and Public Health. Morens DM, Fauci AS. J Infect Dis. 2017 Dec 16;216(suppl_10):S857-S859.
Links:
Video: Uncovering Hidden Zika Outbreaks (Florida Gulf Coast University, Fort Myers)
Zika Virus (National Institute of Allergy and Infectious Diseases/NIH)
Zika Virus Vaccines (NIAID)
Zika Free Florida (Florida Department of Health, Tallahassee)
Grubaugh Lab (Yale School of Public Health, New Haven, CT)
Andersen Lab (The Scripps Research Institute, La Jolla, CA)
NIH Support: National Institute of Allergy and Infectious Diseases; National Center for Advancing Translational Sciences
Protein Links Gut Microbes, Biological Clocks, and Weight Gain
Posted on by Dr. Francis Collins

Caption: Lipids (red) inside mouse intestinal cells with and without NFIL3.
Credit: Lora V. Hooper, University of Texas Southwestern Medical Center, Dallas
The American epidemic of obesity is a major public health concern, and keeping off the extra pounds is a concern for many of us. Yet it can also be a real challenge for people who may eat normally but get their days and nights mixed up, including night-shift workers and those who regularly travel overseas. Why is that?
The most obvious reason is the odd hours throw a person’s 24-hour biological clock—and metabolism—out of sync. But an NIH-funded team of researchers has new evidence in mice to suggest the answer could go deeper to include the trillions of microbes that live in our guts—and, more specifically, the way they “talk” to intestinal cells. Their studies suggest that what gut microbes “say” influences the activity of a key clock-driven protein called NFIL3, which can set intestinal cells up to absorb and store more fat from the diet while operating at hours that might run counter to our fixed biological clocks.
Explaining the Traveler’s First-Night Sleep Problem
Posted on by Dr. Francis Collins

Stock photo/Wavebreakmedia Ltd
This past weekend, I attended a scientific meeting in New York. As often seems to happen to me in a hotel, I tossed and turned and woke up feeling not very rested. The second night I did a bit better. Why is this? Using advanced neuroimaging techniques to study volunteers in a sleep lab, NIH-funded researchers have come up with a biological explanation for this phenomenon, known as “the first-night effect.”
As it turns out, the first night when a person goes to sleep in a new place, a portion of the left hemisphere of his or her brain remains unusually active, apparently to stay alert for any signs of danger. The new findings not only provide important insights into the function of the human brain, they also suggest methods to prevent the first-night effect and thereby help travelers like me in our ongoing quest to get a good night’s sleep.