Somatic Cell Genome Editing
Revolutionizing Technology to Treat Genetic Diseases: The NIH TARGETED Challenge
Posted on by Lawrence Tabak, D.D.S., Ph.D. and Douglas M. Sheeley, Sc.D., NIH Common Fund

Recent scientific advances in the field of genome editing, which enables precise modifications to DNA, have greatly increased the potential to treat genetic diseases. Despite revolutionary progress in this area, treatment options remain limited. Several scientific challenges must be addressed before gene editing can be widely used in the clinic. For example, gene editing tools may cut in unintended areas in addition to the target site, and more research is necessary to understand how these errors affect patients.
Another key challenge is that many organs remain difficult to reach with gene therapies because we do not have adequate ways to deliver gene editing tools to all cells. While efficient delivery technologies exist for some targets, like liver cells, novel and specialized delivery methods designed for specific cell types and locations in the body are needed to ensure genome editing tools can reach sufficient numbers and types of somatic cells to modify DNA safely and effectively. Somatic cell gene therapies target non-reproductive cells, so the changes only affect the person who receives the gene therapy and are not passed down generation to generation.
To address these challenges, NIH launched the TARGETED (Targeted Genome Editor Delivery) Challenge, a multi-phase competition funded through the NIH Common Fund as part of the NIH Somatic Cell Genome Editing (SCGE) Program. SCGE was funded in 2018 to improve the efficacy and specificity of genome editing to help reduce the burden of common and rare diseases caused by genetic changes.
As part of the TARGETED Challenge, research teams will develop technologies for delivering genome editors to somatic cells. NIH will award up to $6 million in prize money across the challenge.
The Challenge is focused on finding delivery systems that can be programmed with biological or chemical tags that correspond to specific target cells and tissues. These tags would direct the delivery systems and the genome editing therapies to the target cells or tissues—like mail being delivered to different zip codes. Such programmable delivery systems would improve gene editing efficacy by targeting diseases at their source and would enhance safety by reducing undesired impacts on other tissues or cells. Ultimately, the development of safe and effective programmable delivery technologies for genome editors that are applicable to multiple diseases would help advance the application of gene editing therapies into the clinic.
The Challenge also is interested in gene editing delivery technologies that can cross the blood-brain barrier (BBB). The BBB protects the brain by blocking harmful substances from entering the fluid of the central nervous system. Unfortunately, it also blocks the uptake of many therapeutics, hindering treatments for brain diseases. While viruses are one of the few approaches that can be used as delivery systems to cross the BBB, they are expensive and difficult to make. Therefore, there is a pressing need for effective non-viral technologies to deliver genome editing machinery across the BBB to a substantial proportion of clinically relevant brain cell types. Such technologies could have broad implications for the treatment of many neurogenetic diseases.
Solutions to both target areas would not only provide proof-of-concept for the delivery of genome editing therapeutics, but they could be adapted to deliver other types of therapies to treat common and rare diseases in general.
The first phase of the Challenge began on May 15, 2023 and will run until October 5, 2023. More information about the Challenge is available on the TARGETED Genome Editor Delivery Challenge website.
Links:
“National Institutes of Health launch TARGETED Challenge,” NIH Common Fund, May 15, 2023
TARGETED Genome Editor Delivery Challenge (NIH Common Fund)
Somatic Cell Genome Editing Program (NIH Common Fund)
NIH Support: The SCGE program is led by the NIH Common Fund, the National Center for Advancing Translational Sciences (NCATS), and the National Institute of Neurological Disorders and Stroke (NINDS). The Brain Research Through Advancing Innovative Neurotechnologies (BRAIN) Initiative and the National Heart, Lung, and Blood Institute (NHLBI) are also contributors to this Challenge.
What A Year It Was for Science Advances!
Posted on by Dr. Francis Collins

At the close of every year, editors and writers at the journal Science review the progress that’s been made in all fields of science—from anthropology to zoology—to select the biggest advance of the past 12 months. In most cases, this Breakthrough of the Year is as tough to predict as the Oscar for Best Picture. Not in 2020. In a year filled with a multitude of challenges posed by the emergence of the deadly coronavirus disease 2019 (COVID-2019), the breakthrough was the development of the first vaccines to protect against this pandemic that’s already claimed the lives of more than 360,000 Americans.
In keeping with its annual tradition, Science also selected nine runner-up breakthroughs. This impressive list includes at least three areas that involved efforts supported by NIH: therapeutic applications of gene editing, basic research understanding HIV, and scientists speaking up for diversity. Here’s a quick rundown of all the pioneering advances in biomedical research, both NIH and non-NIH funded:
Shots of Hope. A lot of things happened in 2020 that were unprecedented. At the top of the list was the rapid development of COVID-19 vaccines. Public and private researchers accomplished in 10 months what normally takes about 8 years to produce two vaccines for public use, with more on the way in 2021. In my more than 25 years at NIH, I’ve never encountered such a willingness among researchers to set aside their other concerns and gather around the same table to get the job done fast, safely, and efficiently for the world.
It’s also pretty amazing that the first two conditionally approved vaccines from Pfizer and Moderna were found to be more than 90 percent effective at protecting people from infection with SARS-CoV-2, the coronavirus that causes COVID-19. Both are innovative messenger RNA (mRNA) vaccines, a new approach to vaccination.
For this type of vaccine, the centerpiece is a small, non-infectious snippet of mRNA that encodes the instructions to make the spike protein that crowns the outer surface of SARS-CoV-2. When the mRNA is injected into a shoulder muscle, cells there will follow the encoded instructions and temporarily make copies of this signature viral protein. As the immune system detects these copies, it spurs the production of antibodies and helps the body remember how to fend off SARS-CoV-2 should the real thing be encountered.
It also can’t be understated that both mRNA vaccines—one developed by Pfizer and the other by Moderna in conjunction with NIH’s National Institute of Allergy and Infectious Diseases—were rigorously evaluated in clinical trials. Detailed data were posted online and discussed in all-day meetings of an FDA Advisory Committee, open to the public. In fact, given the high stakes, the level of review probably was more scientifically rigorous than ever.
First CRISPR Cures: One of the most promising areas of research now underway involves gene editing. These tools, still relatively new, hold the potential to fix gene misspellings—and potentially cure—a wide range of genetic diseases that were once to be out of reach. Much of the research focus has centered on CRISPR/Cas9. This highly precise gene-editing system relies on guide RNA molecules to direct a scissor-like Cas9 enzyme to just the right spot in the genome to cut out or correct a disease-causing misspelling.
In late 2020, a team of researchers in the United States and Europe succeeded for the first time in using CRISPR to treat 10 people with sickle cell disease and transfusion-dependent beta thalassemia. As published in the New England Journal of Medicine, several months after this non-heritable treatment, all patients no longer needed frequent blood transfusions and are living pain free [1].
The researchers tested a one-time treatment in which they removed bone marrow from each patient, modified the blood-forming hematopoietic stem cells outside the body using CRISPR, and then reinfused them into the body. To prepare for receiving the corrected cells, patients were given toxic bone marrow ablation therapy, in order to make room for the corrected cells. The result: the modified stem cells were reprogrammed to switch back to making ample amounts of a healthy form of hemoglobin that their bodies produced in the womb. While the treatment is still risky, complex, and prohibitively expensive, this work is an impressive start for more breakthroughs to come using gene editing technologies. NIH, including its Somatic Cell Genome Editing program, continues to push the technology to accelerate progress and make gene editing cures for many disorders simpler and less toxic.
Scientists Speak Up for Diversity: The year 2020 will be remembered not only for COVID-19, but also for the very public and inescapable evidence of the persistence of racial discrimination in the United States. Triggered by the killing of George Floyd and other similar events, Americans were forced to come to grips with the fact that our society does not provide equal opportunity and justice for all. And that applies to the scientific community as well.
Science thrives in safe, diverse, and inclusive research environments. It suffers when racism and bigotry find a home to stifle diversity—and community for all—in the sciences. For the nation’s leading science institutions, there is a place and a calling to encourage diversity in the scientific workplace and provide the resources to let it flourish to everyone’s benefit.
For those of us at NIH, last year’s peaceful protests and hashtags were noticed and taken to heart. That’s one of the many reasons why we will continue to strengthen our commitment to building a culturally diverse, inclusive workplace. For example, we have established the NIH Equity Committee. It allows for the systematic tracking and evaluation of diversity and inclusion metrics for the intramural research program for each NIH institute and center. There is also the recently founded Distinguished Scholars Program, which aims to increase the diversity of tenure track investigators at NIH. Recently, NIH also announced that it will provide support to institutions to recruit diverse groups or “cohorts” of early-stage research faculty and prepare them to thrive as NIH-funded researchers.
AI Disentangles Protein Folding: Proteins, which are the workhorses of the cell, are made up of long, interconnected strings of amino acids that fold into a wide variety of 3D shapes. Understanding the precise shape of a protein facilitates efforts to figure out its function, its potential role in a disease, and even how to target it with therapies. To gain such understanding, researchers often try to predict a protein’s precise 3D chemical structure using basic principles of physics—including quantum mechanics. But while nature does this in real time zillions of times a day, computational approaches have not been able to do this—until now.
Of the roughly 170,000 proteins mapped so far, most have had their structures deciphered using powerful imaging techniques such as x-ray crystallography and cryo–electron microscopy (cryo-EM). But researchers estimate that there are at least 200 million proteins in nature, and, as amazing as these imaging techniques are, they are laborious, and it can take many months or years to solve 3D structure of a single protein. So, a breakthrough certainly was needed!
In 2020, researchers with the company Deep Mind, London, developed an artificial intelligence (AI) program that rapidly predicts most protein structures as accurately as x-ray crystallography and cryo-EM can map them [2]. The AI program, called AlphaFold, predicts a protein’s structure by computationally modeling the amino acid interactions that govern its 3D shape.
Getting there wasn’t easy. While a complete de novo calculation of protein structure still seemed out of reach, investigators reasoned that they could kick start the modeling if known structures were provided as a training set to the AI program. Utilizing a computer network built around 128 machine learning processors, the AlphaFold system was created by first focusing on the 170,000 proteins with known structures in a reiterative process called deep learning. The process, which is inspired by the way neural networks in the human brain process information, enables computers to look for patterns in large collections of data. In this case, AlphaFold learned to predict the underlying physical structure of a protein within a matter of days. This breakthrough has the potential to accelerate the fields of structural biology and protein research, fueling progress throughout the sciences.
How Elite Controllers Keep HIV at Bay: The term “elite controller” might make some people think of video game whizzes. But here, it refers to the less than 1 percent of people living with human immunodeficiency virus (HIV) who’ve somehow stayed healthy for years without taking antiretroviral drugs. In 2020, a team of NIH-supported researchers figured out why this is so.
In a study of 64 elite controllers, published in the journal Nature, the team discovered a link between their good health and where the virus has inserted itself in their genomes [3]. When a cell transcribes a gene where HIV has settled, this so-called “provirus,” can produce more virus to infect other cells. But if it settles in a part of a chromosome that rarely gets transcribed, sometimes called a gene desert, the provirus is stuck with no way to replicate. Although this discovery won’t cure HIV/AIDS, it points to a new direction for developing better treatment strategies.
In closing, 2020 presented more than its share of personal and social challenges. Among those challenges was a flood of misinformation about COVID-19 that confused and divided many communities and even families. That’s why the editors and writers at Science singled out “a second pandemic of misinformation” as its Breakdown of the Year. This divisiveness should concern all of us greatly, as COVID-19 cases continue to soar around the country and our healthcare gets stretched to the breaking point. I hope and pray that we will all find a way to come together, both in science and in society, as we move forward in 2021.
References:
[1] CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. Frangoul H et al. N Engl J Med. 2020 Dec 5.
[2] ‘The game has changed.’ AI triumphs at protein folding. Service RF. Science. 04 Dec 2020.
[3] Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Jiang C et al. Nature. 2020 Sep;585(7824):261-267.
Links:
COVID-19 Research (NIH)
2020 Science Breakthrough of the Year (American Association for the Advancement of Science, Washington, D.C)
Nano-Sized Solution for Efficient and Versatile CRISPR Gene Editing
Posted on by Dr. Francis Collins

Credit: Guojun Chen and Amr Abdeen, University of Wisconsin-Madison
If used to make non-heritable genetic changes, CRISPR gene-editing technology holds tremendous promise for treating or curing a wide range of devastating disorders, including sickle cell disease, vision loss, and muscular dystrophy. Early efforts to deliver CRISPR-based therapies to affected tissues in a patient’s body typically have involved packing the gene-editing tools into viral vectors, which may cause unwanted immune reactions and other adverse effects.
Now, NIH-supported researchers have developed an alternative CRISPR delivery system: nanocapsules. Not only do these tiny, synthetic capsules appear to pose a lower risk of side effects, they can be precisely customized to deliver their gene-editing payloads to many different types of cells or tissues in the body, which can be extremely tough to do with a virus. Another advantage of these gene-editing nanocapsules is that they can be freeze-dried into a powder that’s easier than viral systems to transport, store, and administer at different doses.
In findings published in Nature Nanotechnology [1], researchers, led by Shaoqin Gong and Krishanu Saha, University of Wisconsin-Madison, developed the nanocapsules with specific design criteria in mind. They would need to be extremely small, about the size of a small virus, for easy entry into cells. Their surface would need to be adaptable for targeting different cell types. They also had to be highly stable in the bloodstream and yet easily degraded to release their contents once inside a cell.
After much hard work in the lab, they created their prototype. It features a thin polymer shell that’s easily decorated with peptides or other ingredients to target the nanocapsule to a predetermined cell type.
At just 25 nanometers in diameter, each nanocapsule still has room to carry cargo. That cargo includes a single CRISPR/Cas9 scissor-like enzyme for snipping DNA and a guide RNA that directs it to the right spot in the genome for editing.
In the bloodstream, the nanocapsules remain fully intact. But, once inside a cell, their polymer shells quickly disintegrate and release the gene-editing payload. How is this possible? The crosslinking molecules that hold the polymer together immediately degrade in the presence of another molecule, called glutathione, which is found at high levels inside cells.
The studies showed that human cells grown in the lab readily engulf and take the gene-editing nanocapsules into bubble-like endosomes. Their gene-editing contents are then released into the cytoplasm where they can begin making their way to a cell’s nucleus within a few hours.
Further study in lab dishes showed that nanocapsule delivery of CRISPR led to precise gene editing of up to about 80 percent of human cells with little sign of toxicity. The gene-editing nanocapsules also retained their potency even after they were freeze-dried and reconstituted.
But would the nanocapsules work in a living system? To find out, the researchers turned to mice, targeting their nanocapsules to skeletal muscle and tissue in the retina at the back of eye. Their studies showed that nanocapsules injected into muscle or the tight subretinal space led to efficient gene editing. In the eye, the nanocapsules worked especially well in editing retinal cells when they were decorated with a chemical ingredient known to bind an important retinal protein.
Based on their initial results, the researchers anticipate that their delivery system could reach most cells and tissues for virtually any gene-editing application. In fact, they are now exploring the potential of their nanocapsules for editing genes within brain tissue.
I’m also pleased to note that Gong and Saha’s team is part of a nationwide consortium on genome editing supported by NIH’s recently launched Somatic Cell Genome Editing program. This program is dedicated to translating breakthroughs in gene editing into treatments for as many genetic diseases as possible. So, we can all look forward to many more advances like this one.
Reference:
[1] A biodegradable nanocapsule delivers a Cas9 ribonucleoprotein complex for in vivo genome editing. Chen G, Abdeen AA, Wang Y, Shahi PK, Robertson S, Xie R, Suzuki M, Pattnaik BR, Saha K, Gong S. Nat Nanotechnol. 2019 Sep 9.
Links:
Somatic Cell Genome Editing (NIH)
Saha Lab (University of Wisconsin-Madison)
Shaoqin (Sarah) Gong (University of Wisconsin-Madison)
NIH Support: National Eye Institute; National Institute of General Medical Sciences; National Institute of Neurological Disorders and Stroke; National Heart, Lung, and Blood Institute; Common Fund
More Progress Toward Gene Editing for Kids with Muscular Dystrophy
Posted on by Dr. Francis Collins

Thanks to CRISPR and other gene editing technologies, hopes have never been greater for treating or even curing Duchenne muscular dystrophy (DMD) and many other rare, genetic diseases that once seemed tragically out of reach. The latest encouraging news comes from a study in which a single infusion of a CRISPR editing system produced lasting benefits in a mouse model of DMD.
There currently is no way to cure DMD, an ultimately fatal disease that mainly affects boys. Caused by mutations in a gene that codes for a critical protein called dystrophin, DMD progressively weakens the skeletal and heart muscles. People with DMD are usually in wheelchairs by the age of 10, with most dying before the age of 30.
The exquisite targeting ability of CRISPR/Cas9 editing systems rely on a sequence-specific guide RNA to direct a scissor-like, bacterial enzyme (Cas9) to just the right spot in the genome, where it can be used to cut out, replace, or repair disease-causing mutations. In previous studies in mice and dogs, researchers directly infused CRISPR systems directly into the animals bodies. This “in vivo” approach to gene editing successfully restored production of functional dystrophin proteins, strengthening animals’ muscles within weeks of treatment.
But an important question remained: would CRISPR’s benefits persist over the long term? The answer in a mouse model of DMD appears to be “yes,” according to findings published recently in Nature Medicine by Charles Gersbach, Duke University, Durham, NC, and his colleagues [1]. Specifically, the NIH-funded team found that after mice with DMD received one infusion of a specially designed CRISPR/Cas9 system, the abnormal gene was edited in a way that restored dystrophin production in skeletal and heart muscles for more than a year. What’s more, lasting improvements were seen in the structure of the animals’ muscles throughout the same time period.
As exciting as these results may be, much more research is needed to explore both the safety and the efficacy of in vivo gene editing before it can be tried in humans with DMD. For instance, the researchers found that older mice that received the editing system developed an immune response to the bacterially-derived Cas9 protein. However, this response didn’t prevent the CRISPR/Cas9 system from doing its job or appear to cause any adverse effects. Interestingly, younger animals didn’t show such a response.
It’s worth noting that the immune systems of mice and people often respond quite differently. But the findings do highlight some possible challenges of such treatments, as well as approaches to reduce possible side effects. For instance, the latest findings suggest CRISPR/Cas9 treatment might best be done early in life, before an infant’s immune system is fully developed. Also, if it’s necessary to deliver CRISPR/Cas9 to older individuals, it may be beneficial to suppress the immune system temporarily.
Another concern about CRISPR technology is the potential for damaging, “off-target” edits to other parts of the genome. In the new work, the Duke team found that its CRISPR system made very few “off-target” edits. However, the system did make a surprising number of complex edits to the targeted dystrophin gene, including integration of the viral vector used to deliver Cas9. While those editing “errors” might reduce the efficacy of treatment, researchers said they didn’t appear to affect the health of the mice studied.
It’s important to emphasize that this gene editing research aimed at curing DMD is being done in non-reproductive (somatic) cells, primarily muscle tissue. The NIH does not support the use of gene editing technologies in human embryos or human reproductive (germline) cells, which would change the genetic makeup of future offspring.
As such, the Duke researchers’ CRISPR/Cas9 system is designed to work optimally in a range of muscle and muscle-progenitor cells. Still, they were able to detect editing of the dystrophin-producing gene in the liver, kidney, brain, and other tissues. Importantly, there was no evidence of edits in the germline cells of the mice. The researchers note that their CRISPR system can be reconfigured to limit gene editing to mature muscle cells, although that may reduce the treatment’s efficacy.
It’s truly encouraging to see that CRISPR gene editing may confer lasting benefits in an animal model of DMD, but a great many questions remain before trying this new approach in kids with DMD. But that time is coming—so let’s boldly go forth and get answers to those questions on behalf of all who are affected by this heartbreaking disease.
Reference:
[1] Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nelson CE, Wu Y, Gemberling MP, Oliver ML, Waller MA, Bohning JD, Robinson-Hamm JN, Bulaklak K, Castellanos Rivera RM, Collier JH, Asokan A, Gersbach CA. Nat Med. 2019 Feb 18.
Links:
Muscular Dystrophy Information Page (National Institute of Neurological Disorders and Stroke/NIH)
Gersbach Lab (Duke University, Durham, NC)
Somatic Cell Genome Editing (Common Fund/NIH)
NIH Support: National Institute of Arthritis and Musculoskeletal and Skin Diseases; National Institute of Biomedical Imaging and Bioengineering
Gene Editing in Dogs Boosts Hope for Kids with Muscular Dystrophy
Posted on by Dr. Francis Collins
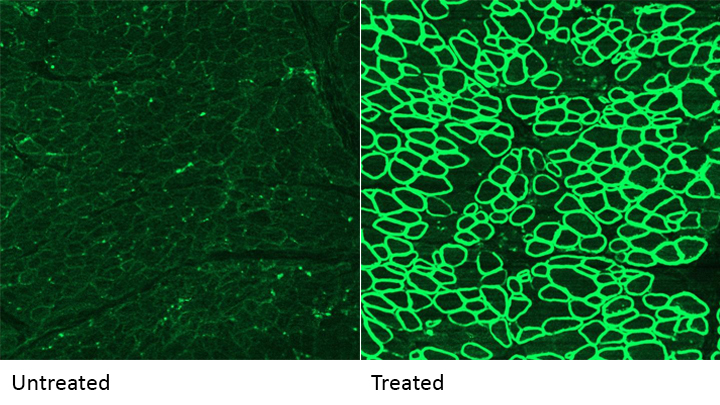
Caption: A CRISPR/cas9 gene editing-based treatment restored production of dystrophin proteins (green) in the diaphragm muscles of dogs with Duchenne muscular dystrophy.
Credit: UT Southwestern
CRISPR and other gene editing tools hold great promise for curing a wide range of devastating conditions caused by misspellings in DNA. Among the many looking to gene editing with hope are kids with Duchenne muscular dystrophy (DMD), an uncommon and tragically fatal genetic disease in which their muscles—including skeletal muscles, the heart, and the main muscle used for breathing—gradually become too weak to function. Such hopes were recently buoyed by a new study that showed infusion of the CRISPR/Cas9 gene editing system could halt disease progression in a dog model of DMD.
As seen in the micrographs above, NIH-funded researchers were able to use the CRISPR/Cas9 editing system to restore production of a critical protein, called dystrophin, by up to 92 percent in the muscle tissue of affected dogs. While more study is needed before clinical trials could begin in humans, this is very exciting news, especially when one considers that boosting dystrophin levels by as little as 15 percent may be enough to provide significant benefit for kids with DMD.