Duchenne muscular dystrophy
Engineering a Better Way to Deliver Therapeutic Genes to Muscles
Posted on by Dr. Francis Collins

Amid all the progress toward ending the COVID-19 pandemic, it’s worth remembering that researchers here and around the world continue to make important advances in tackling many other serious health conditions. As an inspiring NIH-supported example, I’d like to share an advance on the use of gene therapy for treating genetic diseases that progressively degenerate muscle, such as Duchenne muscular dystrophy (DMD).
As published recently in the journal Cell, researchers have developed a promising approach to deliver therapeutic genes and gene editing tools to muscle more efficiently, thus requiring lower doses [1]. In animal studies, the new approach has targeted muscle far more effectively than existing strategies. It offers an exciting way forward to reduce unwanted side effects from off-target delivery, which has hampered the development of gene therapy for many conditions.
In boys born with DMD (it’s an X-linked disease and therefore affects males), skeletal and heart muscles progressively weaken due to mutations in a gene encoding a critical muscle protein called dystrophin. By age 10, most boys require a wheelchair. Sadly, their life expectancy remains less than 30 years.
The hope is gene therapies will one day treat or even cure DMD and allow people with the disease to live longer, high-quality lives. Unfortunately, the benign adeno-associated viruses (AAVs) traditionally used to deliver the healthy intact dystrophin gene into cells mostly end up in the liver—not in muscles. It’s also the case for gene therapy of many other muscle-wasting genetic diseases.
The heavy dose of viral vector to the liver is not without concern. Recently and tragically, there have been deaths in a high-dose AAV gene therapy trial for X-linked myotubular myopathy (XLMTM), a different disorder of skeletal muscle in which there may already be underlying liver disease, potentially increasing susceptibility to toxicity.
To correct this concerning routing error, researchers led by Mohammadsharif Tabebordbar in the lab of Pardis Sabeti, Broad Institute of MIT and Harvard and Harvard University, Cambridge, MA, have now assembled an optimized collection of AAVs. They have been refined to be about 10 times better at reaching muscle fibers than those now used in laboratory studies and clinical trials. In fact, researchers call them myotube AAVs, or MyoAAVs.
MyoAAVs can deliver therapeutic genes to muscle at much lower doses—up to 250 times lower than what’s needed with traditional AAVs. While this approach hasn’t yet been tried in people, animal studies show that MyoAAVs also largely avoid the liver, raising the prospect for more effective gene therapies without the risk of liver damage and other serious side effects.
In the Cell paper, the researchers demonstrate how they generated MyoAAVs, starting out with the commonly used AAV9. Their goal was to modify the outer protein shell, or capsid, to create an AAV that would be much better at specifically targeting muscle. To do so, they turned to their capsid engineering platform known as, appropriately enough, DELIVER. It’s short for Directed Evolution of AAV capsids Leveraging In Vivo Expression of transgene RNA.
Here’s how DELIVER works. The researchers generate millions of different AAV capsids by adding random strings of amino acids to the portion of the AAV9 capsid that binds to cells. They inject those modified AAVs into mice and then sequence the RNA from cells in muscle tissue throughout the body. The researchers want to identify AAVs that not only enter muscle cells but that also successfully deliver therapeutic genes into the nucleus to compensate for the damaged version of the gene.
This search delivered not just one AAV—it produced several related ones, all bearing a unique surface structure that enabled them specifically to target muscle cells. Then, in collaboration with Amy Wagers, Harvard University, Cambridge, MA, the team tested their MyoAAV toolset in animal studies.
The first cargo, however, wasn’t a gene. It was the gene-editing system CRISPR-Cas9. The team found the MyoAAVs correctly delivered the gene-editing system to muscle cells and also repaired dysfunctional copies of the dystrophin gene better than the CRISPR cargo carried by conventional AAVs. Importantly, the muscles of MyoAAV-treated animals also showed greater strength and function.
Next, the researchers teamed up with Alan Beggs, Boston Children’s Hospital, and found that MyoAAV was effective in treating mouse models of XLMTM. This is the very condition mentioned above, in which very high dose gene therapy with a current AAV vector has led to tragic outcomes. XLMTM mice normally die in 10 weeks. But, after receiving MyoAAV carrying a corrective gene, all six mice had a normal lifespan. By comparison, mice treated in the same way with traditional AAV lived only up to 21 weeks of age. What’s more, the researchers used MyoAAV at a dose 100 times lower than that currently used in clinical trials.
While further study is needed before this approach can be tested in people, MyoAAV was also used to successfully introduce therapeutic genes into human cells in the lab. This suggests that the early success in animals might hold up in people. The approach also has promise for developing AAVs with potential for targeting other organs, thereby possibly providing treatment for a wide range of genetic conditions.
The new findings are the result of a decade of work from Tabebordbar, the study’s first author. His tireless work is also personal. His father has a rare genetic muscle disease that has put him in a wheelchair. With this latest advance, the hope is that the next generation of promising gene therapies might soon make its way to the clinic to help Tabebordbar’s father and so many other people.
Reference:
[1] Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Tabebordbar M, Lagerborg KA, Stanton A, King EM, Ye S, Tellez L, Krunnfusz A, Tavakoli S, Widrick JJ, Messemer KA, Troiano EC, Moghadaszadeh B, Peacker BL, Leacock KA, Horwitz N, Beggs AH, Wagers AJ, Sabeti PC. Cell. 2021 Sep 4:S0092-8674(21)01002-3.
Links:
Muscular Dystrophy Information Page (National Institute of Neurological Disorders and Stroke/NIH)
X-linked myotubular myopathy (Genetic and Rare Diseases Information Center/National Center for Advancing Translational Sciences/NIH)
Somatic Cell Genome Editing (Common Fund/NIH)
Mohammadsharif Tabebordbar (Broad Institute of MIT and Harvard and Harvard University, Cambridge, MA)
Sabeti Lab (Broad Institute of MIT and Harvard and Harvard University)
NIH Support: Eunice Kennedy Shriver National Institute of Child Health and Human Development; Common Fund
More Progress Toward Gene Editing for Kids with Muscular Dystrophy
Posted on by Dr. Francis Collins

Thanks to CRISPR and other gene editing technologies, hopes have never been greater for treating or even curing Duchenne muscular dystrophy (DMD) and many other rare, genetic diseases that once seemed tragically out of reach. The latest encouraging news comes from a study in which a single infusion of a CRISPR editing system produced lasting benefits in a mouse model of DMD.
There currently is no way to cure DMD, an ultimately fatal disease that mainly affects boys. Caused by mutations in a gene that codes for a critical protein called dystrophin, DMD progressively weakens the skeletal and heart muscles. People with DMD are usually in wheelchairs by the age of 10, with most dying before the age of 30.
The exquisite targeting ability of CRISPR/Cas9 editing systems rely on a sequence-specific guide RNA to direct a scissor-like, bacterial enzyme (Cas9) to just the right spot in the genome, where it can be used to cut out, replace, or repair disease-causing mutations. In previous studies in mice and dogs, researchers directly infused CRISPR systems directly into the animals bodies. This “in vivo” approach to gene editing successfully restored production of functional dystrophin proteins, strengthening animals’ muscles within weeks of treatment.
But an important question remained: would CRISPR’s benefits persist over the long term? The answer in a mouse model of DMD appears to be “yes,” according to findings published recently in Nature Medicine by Charles Gersbach, Duke University, Durham, NC, and his colleagues [1]. Specifically, the NIH-funded team found that after mice with DMD received one infusion of a specially designed CRISPR/Cas9 system, the abnormal gene was edited in a way that restored dystrophin production in skeletal and heart muscles for more than a year. What’s more, lasting improvements were seen in the structure of the animals’ muscles throughout the same time period.
As exciting as these results may be, much more research is needed to explore both the safety and the efficacy of in vivo gene editing before it can be tried in humans with DMD. For instance, the researchers found that older mice that received the editing system developed an immune response to the bacterially-derived Cas9 protein. However, this response didn’t prevent the CRISPR/Cas9 system from doing its job or appear to cause any adverse effects. Interestingly, younger animals didn’t show such a response.
It’s worth noting that the immune systems of mice and people often respond quite differently. But the findings do highlight some possible challenges of such treatments, as well as approaches to reduce possible side effects. For instance, the latest findings suggest CRISPR/Cas9 treatment might best be done early in life, before an infant’s immune system is fully developed. Also, if it’s necessary to deliver CRISPR/Cas9 to older individuals, it may be beneficial to suppress the immune system temporarily.
Another concern about CRISPR technology is the potential for damaging, “off-target” edits to other parts of the genome. In the new work, the Duke team found that its CRISPR system made very few “off-target” edits. However, the system did make a surprising number of complex edits to the targeted dystrophin gene, including integration of the viral vector used to deliver Cas9. While those editing “errors” might reduce the efficacy of treatment, researchers said they didn’t appear to affect the health of the mice studied.
It’s important to emphasize that this gene editing research aimed at curing DMD is being done in non-reproductive (somatic) cells, primarily muscle tissue. The NIH does not support the use of gene editing technologies in human embryos or human reproductive (germline) cells, which would change the genetic makeup of future offspring.
As such, the Duke researchers’ CRISPR/Cas9 system is designed to work optimally in a range of muscle and muscle-progenitor cells. Still, they were able to detect editing of the dystrophin-producing gene in the liver, kidney, brain, and other tissues. Importantly, there was no evidence of edits in the germline cells of the mice. The researchers note that their CRISPR system can be reconfigured to limit gene editing to mature muscle cells, although that may reduce the treatment’s efficacy.
It’s truly encouraging to see that CRISPR gene editing may confer lasting benefits in an animal model of DMD, but a great many questions remain before trying this new approach in kids with DMD. But that time is coming—so let’s boldly go forth and get answers to those questions on behalf of all who are affected by this heartbreaking disease.
Reference:
[1] Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nelson CE, Wu Y, Gemberling MP, Oliver ML, Waller MA, Bohning JD, Robinson-Hamm JN, Bulaklak K, Castellanos Rivera RM, Collier JH, Asokan A, Gersbach CA. Nat Med. 2019 Feb 18.
Links:
Muscular Dystrophy Information Page (National Institute of Neurological Disorders and Stroke/NIH)
Gersbach Lab (Duke University, Durham, NC)
Somatic Cell Genome Editing (Common Fund/NIH)
NIH Support: National Institute of Arthritis and Musculoskeletal and Skin Diseases; National Institute of Biomedical Imaging and Bioengineering
Accelerating Cures in the Genomic Age: The Sickle Cell Example
Posted on by Dr. Francis Collins

Forty-five years ago, when I was a first-year medical student, a lecturer introduced me to a young man with sickle cell disease (SCD). Sickle cell disease is the first “molecular disease”, with its cause having been identified decades ago. That helped me see the connection between the abstract concepts of molecular genetics and their real-world human consequences in a way no textbook could. In fact, it inspired some of my earliest research on human hemoglobin disorders, which I conducted as a postdoctoral fellow.
Today, I’m heartened to report that, thanks to decades of biomedical advances, we stand on the verge of a cure for SCD. While at the American Society of Hematology meeting in San Diego last week, I was excited to be part of a discussion about how the tools and technologies arising from the Human Genome Project are accelerating the quest for cures.
The good news at the meeting included some promising, early results from human clinical trials of SCD gene therapies, including new data from the NIH Clinical Center. Researchers also presented very encouraging pre-clinical work on how gene-editing technologies, such as CRISPR, can be used in ways that may open the door to curing everyone with SCD. In fact, just days before the meeting, the first clinical trial for a CRISPR approach to SCD opened.
One important note: the gene editing research aimed at curing SCD is being done in non-reproductive (somatic) cells. The NIH does not support the use of gene editing technologies in human embryos (germline). I recently reiterated our opposition to germline gene editing, in response to an unethical experiment by a researcher in China who claims to have used CRISPR editing on embryos to produce twin girls resistant to HIV.
SCD affects approximately 100,000 people in the United States, and another 20 million worldwide, mostly in developing nations. This inherited, potentially life-threatening disorder is caused by a specific point mutation in a gene that codes for the beta chain of hemoglobin, a molecule found in red blood cells that deliver oxygen throughout the body. In people with SCD, the mutant hemoglobin forms insoluble aggregates when de-oxygenated. As a result the red cells assume a sickle shape, rather than the usual donut shape. These sickled cells clump together and stick in small blood vessels, resulting in severe pain, blood cell destruction, anemia, stroke, pulmonary hypertension, organ failure, and much too often, early death.
The need for a widespread cure for SCD is great. Since 1998, doctors have used a drug called hydroxyurea to reduce symptoms, but it can cause serious side effects and increase the risk of certain cancers. Blood transfusions can also ease symptoms in certain instances, but they too come with risks and complications. At the present time, the only way to cure SCD is a bone marrow transplant. However, transplants are not an option for many patients due to lack of matched marrow donors.
The good news is that novel genetic approaches have raised hopes of a widespread cure for SCD, possibly even within five to 10 years. So, in September, NIH’s National Heart, Lung, and Blood Institute launched the Cure Sickle Cell Initiative to accelerate development of the most promising of these next generation of therapies
At the ASH meeting, that first wave of this progress was evident. A team led by NHLBI’s John Tisdale, in collaboration with Bluebird bio, Cambridge, MA, was among the groups that presented impressive early results from human clinical trials testing novel gene replacement therapies for SCD. In the NIH trial, researchers removed blood precursor cells, called hematopoietic stem cells (HSCs), from a patient’s own bone marrow or bloodstream and used a harmless virus to insert a sickle-resistant hemoglobin gene. Then, after a chemotherapy infusion to condition the patient’s existing bone marrow, they returned the corrected cells to the patient.
So far, nine SCD patients have received the most advanced form of the experimental gene therapy, and Tisdale presented data on those who were farthest out from treatment [1,2]. His team found that in the four patients who were at least six months out, levels of gene therapy-derived hemoglobin were found to equal or exceed their levels of SCD hemoglobin.
Very cool science, but what does this mean for SCD patients’ health and well-being? Well, none of the gene therapy trial participants have required a blood transfusion during the follow-up period. In addition, improvements were seen in their hemoglobin levels and key markers of blood-cell destruction (total bilirubin concentration, lactate dehydrogenase, and reticulocyte counts) compared to baseline. Most importantly, in the years leading up to the clinical trial, all of the participants had experienced frequent painful sickle crises, in which sickled cells blocked their blood vessels. No such episodes were reported among the participants in the months after they received the gene therapy.
Researchers did report that one patient receiving this form of gene therapy developed myelodysplastic syndrome (MDS), a serious condition in which the blood-forming cells in the bone marrow become abnormal. However, there is no indication that the gene replacement technology itself caused the problem, and MDS has previously been linked to the chemotherapy drugs used in conditioning regimens before bone marrow transplants.
The NIH trial is just one of several clinical trials for SCD that are using viral vectors to deliver a variety of genes with therapeutic potential. Other trials actively recruiting are led by researchers at Boston Children’s Hospital, Cincinnati Children’s Hospital, and the University of California, Los Angeles.
While it’s hoped that genes inserted by viral vectors will provide long-lasting or curative treatment, other researchers are betting that new gene-editing technologies, such as CRISPR, will offer the best chance for developing a widespread cure for SCD. One strategy being eyed by these “gene editors” is to correct the SCD mutation, replacing it with a normal gene. Another strategy involves knocking out certain DNA sequences to reactivate production of fetal hemoglobin (HbF).
The HbF protein is produced in the developing fetus to give it better access to oxygen from the mother’s bloodstream. But shortly after birth, the production of fetal hemoglobin shuts down, and the adult form kicks in. Adults normally have very low levels of fetal hemoglobin, which makes sense. However, from genome-wide association studies of human genetic variation, we know that that actual levels of HbF are under genetic control.
A major factor has been mapped to the BCL11A gene, which has subsequently been found to be a master mediator for the fetal to adult hemoglobin switch. Specifically, variations in a red cell specific enhancer of BCL11A affect an adult’s level of HbF— levels of BCL11A protein lead to higher amounts of fetal hemoglobin. Furthermore, it’s been known for some time that rare individuals keep on producing relatively high levels of hemoglobin into adulthood. If people with SCD happen to have a rare mutation that keeps fetal hemoglobin production active in adulthood (the first of these was found as part of my postdoctoral research), their SCD symptoms are much less severe.
Currently, two groups—CRISPR Therapeutics/Vertex Pharmaceuticals and Sangamo Therapeutics/Bioverativ—are gearing up to begin the first U.S. human clinical trials of gene-editing for SCD within the next few months. While they employ different technologies, both approaches involve removing a patient’s HSCs, using gene editing to knock out the BCL11A red cell enhancer, and then returning the gene-edited cells to the patient. The hope is that the gene-edited cells will greatly boost fetal hemoglobin production, thereby offsetting the effects of SCD.
All of this is exciting news for the 100,000 people living in the United States who have SCD. But what about the 300,000 babies born with SCD every year in other parts of the world, mostly in low- and middle-income countries?
The complicated, high-tech procedures that I just described may not be practical for a very long time in places like sub-Saharan Africa. That’s one reason why NIH recently launched a new effort to speed the development of safe, effective genome-editing approaches that could be delivered directly into a patient’s body (in vivo), perhaps by infusion of the CRISPR gene editing apparatus. Recent preclinical experiments demonstrating the promise of in vivo gene editing for Duchenne muscular dystrophy make me optimistic that NIH’s Somatic Cell Genome Editing Program, which is hosting its first gathering of investigators this week, will be able to develop similar approaches for SCD and many other conditions.
While moving forward in this fast-paced field, it is important that we remain ethical, but also remain bold on behalf of the millions of patients with genetic diseases who are still waiting for a cure. We must continue to assess and address the very serious ethical concerns raised by germline gene editing of human embryos, which will irreversibly alter the DNA instruction book of future children and affect future generations. I continue to argue that we are not ready to undertake such experiments.
But the use of gene editing to treat, perhaps even to cure, children and adults with genetic diseases, by correcting the mutation in their relevant tissues (so-called somatic cell gene editing), without risk of passing those changes on to a future generation, holds enormous promise. Somatic cell gene editing is associated with ethical issues that are much more in line with decades of deep thinking about benefits and risks of therapeutic trials.
Finally, we must recognize that somatic cell gene editing is a profoundly promising approach not only for people with SCD, but for all who are struggling with the thousands of diseases that still have no treatments or cures. Real hope for cures has never been greater.
References:
[1] NIH researcher presents encouraging results for gene therapy for severe sickle cell disease. NIH News Release. December 4, 2018
[2] Bluebird bio presents new data for LentiGlobin gene therapy in sickle cell disease at 60th annual meeting of the American Society of Hematology. Bluebird bio. December 3, 2018
Links:
Sickle Cell Disease (National Heart, Lung, and Blood Institute/NIH)
Cure Sickle Cell Initiative (NHLBI)
John Tisdale (NHLBI)
Somatic Cell Genome Editing Program (Common Fund/NIH)
What are genome editing and CRISPR-Cas9? (National Library of Medicine/NIH)
ClinicalTrials.gov (NIH)
NIH Support: National Heart, Lung, and Blood Institute; Common Fund
Gene Editing in Dogs Boosts Hope for Kids with Muscular Dystrophy
Posted on by Dr. Francis Collins
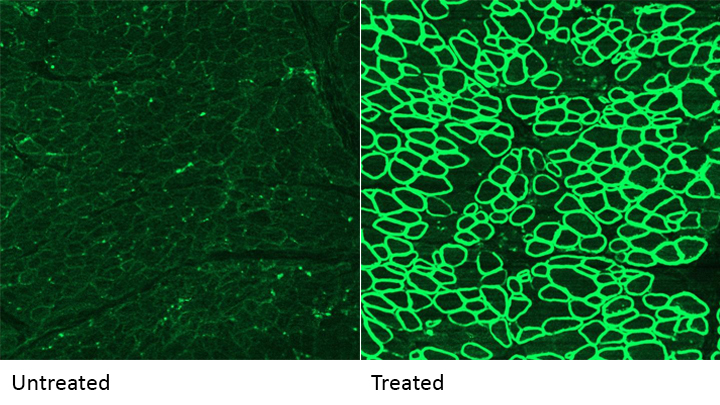
Caption: A CRISPR/cas9 gene editing-based treatment restored production of dystrophin proteins (green) in the diaphragm muscles of dogs with Duchenne muscular dystrophy.
Credit: UT Southwestern
CRISPR and other gene editing tools hold great promise for curing a wide range of devastating conditions caused by misspellings in DNA. Among the many looking to gene editing with hope are kids with Duchenne muscular dystrophy (DMD), an uncommon and tragically fatal genetic disease in which their muscles—including skeletal muscles, the heart, and the main muscle used for breathing—gradually become too weak to function. Such hopes were recently buoyed by a new study that showed infusion of the CRISPR/Cas9 gene editing system could halt disease progression in a dog model of DMD.
As seen in the micrographs above, NIH-funded researchers were able to use the CRISPR/Cas9 editing system to restore production of a critical protein, called dystrophin, by up to 92 percent in the muscle tissue of affected dogs. While more study is needed before clinical trials could begin in humans, this is very exciting news, especially when one considers that boosting dystrophin levels by as little as 15 percent may be enough to provide significant benefit for kids with DMD.
Gene Editing: Gold Nanoparticle Delivery Shows Promise
Posted on by Dr. Francis Collins
 About a month ago, I had the pleasure of welcoming the Juip (pronounced “Yipe”) family from Michigan to NIH. Although you’d never guess it from this photo, two of the Juip’s five children—9-year-old Claire and 11-year-old Jake (both to my left)—have a rare genetic disease called Friedreich’s ataxia (FA). This inherited condition causes progressive damage to their nervous systems and their hearts. No treatment currently exists for kids like Claire and Jake, yet this remarkable family has turned this serious health challenge into an opportunity to raise awareness about the need for biomedical research.
About a month ago, I had the pleasure of welcoming the Juip (pronounced “Yipe”) family from Michigan to NIH. Although you’d never guess it from this photo, two of the Juip’s five children—9-year-old Claire and 11-year-old Jake (both to my left)—have a rare genetic disease called Friedreich’s ataxia (FA). This inherited condition causes progressive damage to their nervous systems and their hearts. No treatment currently exists for kids like Claire and Jake, yet this remarkable family has turned this serious health challenge into an opportunity to raise awareness about the need for biomedical research.
One thing that helps keep the Juips optimistic is the therapeutic potential of CRISPR/Cas9, an innovative gene editing system that may someday make it possible to correct the genetic mutations responsible for FA and many other conditions. So, I’m sure the Juips were among those encouraged by the recent news that NIH-funded researchers have developed a highly versatile approach to CRISPR/Cas9-based therapies. Instead of relying on viruses to carry the gene-editing system into cells, the new approach uses tiny particles of gold as the delivery system!