hematology
A CRISPR Approach to Treating Sickle Cell
Posted on by Dr. Francis Collins

Recently, CBS’s “60 Minutes” highlighted the story of Jennelle Stephenson, a brave young woman with sickle cell disease (SCD). Jennelle now appears potentially cured of this devastating condition, thanks to an experimental gene therapy being tested at the NIH Clinical Center in Bethesda, MD. As groundbreaking as this research may be, it’s among a variety of innovative strategies now being tried to cure SCD and other genetic diseases that have long seemed out of reach.
One particularly exciting approach involves using gene editing to increase levels of fetal hemoglobin (HbF) in the red blood cells of people with SCD. Shortly after birth, babies usually stop producing HbF, and switch over to the adult form of hemoglobin. But rare individuals continue to make high levels of HbF throughout their lives. This is referred to as hereditary persistence of fetal hemoglobin (HPFH). (My own postdoctoral research in the early 1980s discovered some of the naturally occurring DNA mutations that lead to this condition.)
Individuals with HPFH are entirely healthy. Strikingly, rare individuals with SCD who also have HPFH have an extremely mild version of sickle cell disease—essentially the presence of significant quantities of HbF provides protection against sickling. So, researchers have been exploring ways to boost HbF in everyone with SCD—and gene editing may provide an effective, long-lasting way to do this.
Clinical trials of this approach are already underway. And new findings reported in Nature Medicine show it may be possible to make the desired edits even more efficiently, raising the possibility that a single infusion of gene-edited cells might be able to cure SCD [1].
Sickle cell disease is caused by a specific point mutation in a gene that codes for the beta chain of hemoglobin. People with just one copy of this mutation have sickle cell trait and are generally healthy. But those who inherit two mutant copies of this gene suffer lifelong consequences of the presence of this abnormal protein. Their red blood cells—normally flexible and donut-shaped—assume the sickled shape that gives SCD its name. The sickled cells clump together and stick in small blood vessels, resulting in severe pain, anemia, stroke, pulmonary hypertension, organ failure, and far too often, early death.
Eleven years ago, a team led by Vijay Sankaran and Stuart Orkin at Boston Children’s Hospital and the Dana-Farber Cancer Institute discovered that a protein called BCL11A seemed to determine HbF levels [2]. Subsequent work showed the protein actually works as a master mediator of the switch from fetal to adult hemoglobin, which normally occurs shortly after birth.
Five years ago, Orkin and Daniel Bauer identified a specific enhancer of BCL11A expression that could be an attractive target for gene editing [3]. They could knock out the enhancer in the bone marrow, and BCL11A would not be produced, allowing HbF to stay switched on.
Because the BCL11A protein is required to turn off production of HbF in red cells. the researchers had another idea. They thought it might be possible to keep HbF on permanently by disrupting BCL11A in blood-forming hematopoietic stem cells (HSCs). The hope was that such a treatment might offer people with SCD a permanent supply of healthy red blood cells.
Fast-forward to the present, and researchers are now testing the ability of gene editing tools to cure the disease. A favorite editing system is CRISPR, which I’ve highlighted on my blog.
CRISPR is a highly precise gene-editing tool that relies on guide RNA molecules to direct a scissor-like Cas9 enzyme to just the right spot in the genome to correct the misspelling. The gene-editing treatment involves removing bone marrow from a patient, modifying the HSCs outside the body using CRISPR gene-editing tools, and then returning them back to the patient. Preclinical studies had shown that CRISPR can be effective in editing BCL11A to boost HbF production.
But questions lingered about the editing efficiency in HSCs versus more common, shorter-lived progenitor cells found in bone marrow samples. The efficiency greatly influences how long the edited cells might benefit patients. Bauer’s team saw room for improvement and, as the new study shows, they were right.
To produce lasting HbF production, it’s important to edit as many HSCs as possible. But it turns out that HSCs are more resistant to editing than other types of cells in bone marrow. With a series of adjustments to the gene-editing protocol, including use of an optimized version of the Cas9 protein, the researchers showed they could push the number of edited genes from about 80 percent to about 95 percent.
Their studies show that the most frequent Cas9 edits in HSCs are tiny insertions of a single DNA “letter.” With that slight edit to the BCL11A gene, HSCs reprogram themselves in a way that ensures long-term HbF production.
As a first test of their CRISPR-edited human HSCs, the researchers carried out the editing on HSCs derived from patients with SCD. Then they transferred the editing cells into immune-compromised mice. Four months later, the mice continued to produce red blood cells that produced high levels of HbF and resisted sickling. Bauer says they’re already taking steps to begin testing cells edited with their optimized protocol in a clinical trial.
What’s truly exciting is that the first U.S. human clinical trials of such a gene-editing approach for SCD are already underway, led by CRISPR Therapeutics/Vertex Pharmaceuticals and Sangamo Therapeutics/Sanofi. In January, CRISPR Therapeutics/Vertex Pharmaceuticals announced that the U.S. Food and Drug Administration (FDA) had granted Fast Track Designation for their CRISPR-based treatment called CTX001 [4].
In that recent “60 Minutes” segment, I dared to suggest that we now have what looks like a cure for SCD. As shown by this new work and the clinical trials underway, we in fact may soon have multiple different strategies to provide cures for this devastating disease. And if this can work for sickle cell, a similar strategy might work for other genetic conditions that currently lack any effective treatment.
References:
[1] Highly efficient therapeutic gene editing of human hematopoietic stem cells. Wu Y, Zeng J, Roscoe BP, Liu P, Yao Q, Lazzarotto CR, Clement K, Cole MA, Luk K, Baricordi C, Shen AH, Ren C, Esrick EB, Manis JP, Dorfman DM, Williams DA, Biffi A, Brugnara C, Biasco L, Brendel C, Pinello L, Tsai SQ, Wolfe SA, Bauer DE. Nat Med. 2019 Mar 25.
[2] Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, Mikkola HK, Hirschhorn JN, Cantor AB, Orkin SH.Science. 2008 Dec 19;322(5909):1839-1842.
[3] An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Bauer DE, Kamran SC, Lessard S, Xu J, Fujiwara Y, Lin C, Shao Z, Canver MC, Smith EC, Pinello L, Sabo PJ, Vierstra J, Voit RA, Yuan GC, Porteus MH, Stamatoyannopoulos JA, Lettre G, Orkin SH. Science. 2013 Oct 11;342(6155):253-257.
[4] CRISPR Therapeutics and Vertex Announce FDA Fast Track Designation for CTX001 for the Treatment of Sickle Cell Disease, CRISPR Therapeutics News Release, Jan. 4, 2019.
Links:
Sickle Cell Disease (National Heart, Lung, and Blood Institute/NIH)
Cure Sickle Cell Initiative (NHLBI)
What are Genome Editing and CRISPR-Cas9? (National Library of Medicine/NIH)
Could Gene Therapy Cure Sickle Cell Anemia? (CBS News)
Daniel Bauer (Dana-Farber Cancer Institute, Boston)
Somatic Cell Genome Editing Program (Common Fund/NIH)
NIH Support: National Heart, Lung, and Blood Institute; National Institute of General Medical Sciences; National Institute of Allergy and Infectious Diseases; National Institute of Diabetes and Digestive and Kidney Diseases
Precision Medicine: Making Warfarin Safer
Posted on by Dr. Francis Collins

Caption: Finding the right dose of the drug warfarin can be tricky, even with this standard test to measure how fast a person’s blood clots.
Credit: Thinkstock/jarun011
Every year, thousands of older Americans require emergency treatment to stop bleeding caused by taking warfarin, a frequently prescribed blood-thinning pill. My own mother received this drug in her later years, and her doctors encountered significant challenges getting the dose right. The problem is too much warfarin causes potentially serious bleeding, while too little leaves those who need the drug vulnerable to developing life-threatening clots in their legs or heart. The difference between too little and too much is distressingly small. But what if before writing a prescription, doctors could test for known genetic markers to help them gauge the amount of warfarin that a person should take?
Such tests have been available to doctors and patients for a few years, but they have not been widely used. The recent results of a national clinical trial offer some of the most convincing evidence that it’s time for that to change. In this study of 1,650 older adults undergoing elective hip or knee surgery, patients whose genetic makeup was used to help determine their dose of warfarin were less likely to suffer adverse events, including major bleeding. This trial marks an encouraging success story for the emerging field of pharmacogenomics, the study of how the variations in our genes affect our responses to medicines.
Sickle Cell Disease: Gene-Editing Tools Point to Possible Ultimate Cure
Posted on by Dr. Francis Collins
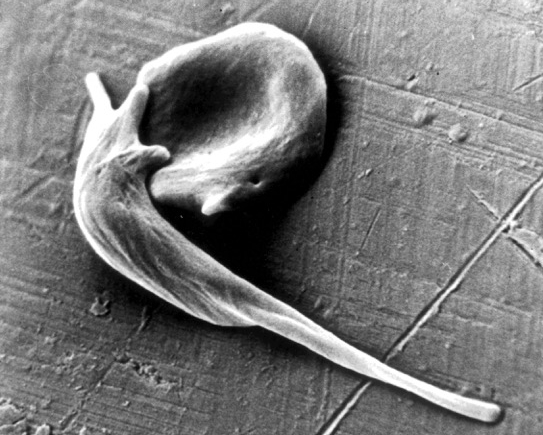
Caption: An electron micrograph showing two red blood cells deformed by crystalline hemoglobin into different “sickle” shapes characteristic of people with sickle cell disease.
Credit: Frans Kuypers: RBClab.com, UCSF Benioff Children’s Hospital Oakland
Scientists first described the sickle-shaped red blood cells that give sickle cell disease its name more than a century ago. By the 1950s, the precise molecular and genetic underpinnings of this painful and debilitating condition had become clear, making sickle cell the first “molecular disease” ever characterized. The cause is a single letter “typo” in the gene encoding oxygen-carrying hemoglobin. Red blood cells containing the defective hemoglobin become stiff, deformed, and prone to clumping. Individuals carrying one copy of the sickle mutation have sickle trait, and are generally fine. Those with two copies have sickle cell disease and face major medical challenges. Yet, despite all this progress in scientific understanding, nearly 70 years later, we still have no safe and reliable means for a cure.
Recent advances in CRISPR/Cas9 gene-editing tools, which the blog has highlighted in the past, have renewed hope that it might be possible to cure sickle cell disease by correcting DNA typos in just the right set of cells. Now, in a study published in Science Translational Medicine, an NIH-funded research team has taken an encouraging step toward this goal [1]. For the first time, the scientists showed that it’s possible to correct the hemoglobin mutation in blood-forming human stem cells, taken directly from donors, at a frequency that might be sufficient to help patients. In addition, their gene-edited human stem cells persisted for 16 weeks when transplanted into mice, suggesting that the treatment might also be long lasting or possibly even curative.