53 Search Results for "Gene Editing"
Engineering a Better Way to Deliver Therapeutic Genes to Muscles
Posted on by Dr. Francis Collins

Amid all the progress toward ending the COVID-19 pandemic, it’s worth remembering that researchers here and around the world continue to make important advances in tackling many other serious health conditions. As an inspiring NIH-supported example, I’d like to share an advance on the use of gene therapy for treating genetic diseases that progressively degenerate muscle, such as Duchenne muscular dystrophy (DMD).
As published recently in the journal Cell, researchers have developed a promising approach to deliver therapeutic genes and gene editing tools to muscle more efficiently, thus requiring lower doses [1]. In animal studies, the new approach has targeted muscle far more effectively than existing strategies. It offers an exciting way forward to reduce unwanted side effects from off-target delivery, which has hampered the development of gene therapy for many conditions.
In boys born with DMD (it’s an X-linked disease and therefore affects males), skeletal and heart muscles progressively weaken due to mutations in a gene encoding a critical muscle protein called dystrophin. By age 10, most boys require a wheelchair. Sadly, their life expectancy remains less than 30 years.
The hope is gene therapies will one day treat or even cure DMD and allow people with the disease to live longer, high-quality lives. Unfortunately, the benign adeno-associated viruses (AAVs) traditionally used to deliver the healthy intact dystrophin gene into cells mostly end up in the liver—not in muscles. It’s also the case for gene therapy of many other muscle-wasting genetic diseases.
The heavy dose of viral vector to the liver is not without concern. Recently and tragically, there have been deaths in a high-dose AAV gene therapy trial for X-linked myotubular myopathy (XLMTM), a different disorder of skeletal muscle in which there may already be underlying liver disease, potentially increasing susceptibility to toxicity.
To correct this concerning routing error, researchers led by Mohammadsharif Tabebordbar in the lab of Pardis Sabeti, Broad Institute of MIT and Harvard and Harvard University, Cambridge, MA, have now assembled an optimized collection of AAVs. They have been refined to be about 10 times better at reaching muscle fibers than those now used in laboratory studies and clinical trials. In fact, researchers call them myotube AAVs, or MyoAAVs.
MyoAAVs can deliver therapeutic genes to muscle at much lower doses—up to 250 times lower than what’s needed with traditional AAVs. While this approach hasn’t yet been tried in people, animal studies show that MyoAAVs also largely avoid the liver, raising the prospect for more effective gene therapies without the risk of liver damage and other serious side effects.
In the Cell paper, the researchers demonstrate how they generated MyoAAVs, starting out with the commonly used AAV9. Their goal was to modify the outer protein shell, or capsid, to create an AAV that would be much better at specifically targeting muscle. To do so, they turned to their capsid engineering platform known as, appropriately enough, DELIVER. It’s short for Directed Evolution of AAV capsids Leveraging In Vivo Expression of transgene RNA.
Here’s how DELIVER works. The researchers generate millions of different AAV capsids by adding random strings of amino acids to the portion of the AAV9 capsid that binds to cells. They inject those modified AAVs into mice and then sequence the RNA from cells in muscle tissue throughout the body. The researchers want to identify AAVs that not only enter muscle cells but that also successfully deliver therapeutic genes into the nucleus to compensate for the damaged version of the gene.
This search delivered not just one AAV—it produced several related ones, all bearing a unique surface structure that enabled them specifically to target muscle cells. Then, in collaboration with Amy Wagers, Harvard University, Cambridge, MA, the team tested their MyoAAV toolset in animal studies.
The first cargo, however, wasn’t a gene. It was the gene-editing system CRISPR-Cas9. The team found the MyoAAVs correctly delivered the gene-editing system to muscle cells and also repaired dysfunctional copies of the dystrophin gene better than the CRISPR cargo carried by conventional AAVs. Importantly, the muscles of MyoAAV-treated animals also showed greater strength and function.
Next, the researchers teamed up with Alan Beggs, Boston Children’s Hospital, and found that MyoAAV was effective in treating mouse models of XLMTM. This is the very condition mentioned above, in which very high dose gene therapy with a current AAV vector has led to tragic outcomes. XLMTM mice normally die in 10 weeks. But, after receiving MyoAAV carrying a corrective gene, all six mice had a normal lifespan. By comparison, mice treated in the same way with traditional AAV lived only up to 21 weeks of age. What’s more, the researchers used MyoAAV at a dose 100 times lower than that currently used in clinical trials.
While further study is needed before this approach can be tested in people, MyoAAV was also used to successfully introduce therapeutic genes into human cells in the lab. This suggests that the early success in animals might hold up in people. The approach also has promise for developing AAVs with potential for targeting other organs, thereby possibly providing treatment for a wide range of genetic conditions.
The new findings are the result of a decade of work from Tabebordbar, the study’s first author. His tireless work is also personal. His father has a rare genetic muscle disease that has put him in a wheelchair. With this latest advance, the hope is that the next generation of promising gene therapies might soon make its way to the clinic to help Tabebordbar’s father and so many other people.
Reference:
[1] Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Tabebordbar M, Lagerborg KA, Stanton A, King EM, Ye S, Tellez L, Krunnfusz A, Tavakoli S, Widrick JJ, Messemer KA, Troiano EC, Moghadaszadeh B, Peacker BL, Leacock KA, Horwitz N, Beggs AH, Wagers AJ, Sabeti PC. Cell. 2021 Sep 4:S0092-8674(21)01002-3.
Links:
Muscular Dystrophy Information Page (National Institute of Neurological Disorders and Stroke/NIH)
X-linked myotubular myopathy (Genetic and Rare Diseases Information Center/National Center for Advancing Translational Sciences/NIH)
Somatic Cell Genome Editing (Common Fund/NIH)
Mohammadsharif Tabebordbar (Broad Institute of MIT and Harvard and Harvard University, Cambridge, MA)
Sabeti Lab (Broad Institute of MIT and Harvard and Harvard University)
NIH Support: Eunice Kennedy Shriver National Institute of Child Health and Human Development; Common Fund
DNA Base Editing May Treat Progeria, Study in Mice Shows
Posted on by Dr. Francis Collins

My good friend Sam Berns was born with a rare genetic condition that causes rapid premature aging. Though Sam passed away in his teens from complications of this condition, called Hutchinson-Gilford progeria syndrome, he’s remembered today for his truly positive outlook on life. Sam expressed it, in part, by his willingness to make adjustments that allowed him, in his words, to put things that he always wanted to do in the “can do” category.
In this same spirit on behalf of the several hundred kids worldwide with progeria and their families, a research collaboration, including my NIH lab, has now achieved a key technical advance to move non-heritable gene editing another step closer to the “can do” category to treat progeria. As published in the journal Nature, our team took advantage of new gene-editing tools to correct for the first time a single genetic misspelling responsible for progeria in a mouse model, with dramatically beneficial effects [1, 2]. This work also has implications for correcting similar single-base typos that cause other inherited genetic disorders.
The outcome of this work is incredibly gratifying for me. In 2003, my NIH lab discovered the DNA mutation that causes progeria. One seemingly small glitch—swapping a “T” in place of a “C” in a gene called lamin A (LMNA)—leads to the production of a toxic protein now known as progerin. Without treatment, children with progeria develop normally intellectually but age at an exceedingly rapid pace, usually dying prematurely from heart attacks or strokes in their early teens.
The discovery raised the possibility that correcting this single-letter typo might one day help or even cure children with progeria. But back then, we lacked the needed tools to edit DNA safely and precisely. To be honest, I didn’t think that would be possible in my lifetime. Now, thanks to advances in basic genomic research, including work that led to the 2020 Nobel Prize in Chemistry, that’s changed. In fact, there’s been substantial progress toward using gene-editing technologies, such as the CRISPR editing system, for treating or even curing a wide range of devastating genetic conditions, such as sickle cell disease and muscular dystrophy
It turns out that the original CRISPR system, as powerful as it is, works better at knocking out genes than correcting them. That’s what makes some more recently developed DNA editing agents and approaches so important. One of them, which was developed by David R. Liu, Broad Institute of MIT and Harvard, Cambridge, MA, and his lab members, is key to these latest findings on progeria, reported by a team including my lab in NIH’s National Human Genome Research Institute and Jonathan Brown, Vanderbilt University Medical Center, Nashville, TN.
The relatively new gene-editing system moves beyond knock-outs to knock-ins [3,4]. Here’s how it works: Instead of cutting DNA as CRISPR does, base editors directly convert one DNA letter to another by enzymatically changing one DNA base to become a different base. The result is much like the find-and-replace function used to fix a typo in a word processor. What’s more, the gene editor does this without cutting the DNA.
Our three labs (Liu, Brown, and Collins) first teamed up with the Progeria Research Foundation, Peabody, MA, to obtain skin cells from kids with progeria. In lab studies, we found that base editors, targeted by an appropriate RNA guide, could successfully correct the LMNA gene in those connective tissue cells. The treatment converted the mutation back to the normal gene sequence in an impressive 90 percent of the cells.
But would it work in a living animal? To get the answer, we delivered a single injection of the DNA-editing apparatus into nearly a dozen mice either three or 14 days after birth, which corresponds in maturation level roughly to a 1-year-old or 5-year-old human. To ensure the findings in mice would be as relevant as possible to a future treatment for use in humans, we took advantage of a mouse model of progeria developed in my NIH lab in which the mice carry two copies of the human LMNA gene variant that causes the condition. Those mice develop nearly all of the features of the human illness
In the live mice, the base-editing treatment successfully edited in the gene’s healthy DNA sequence in 20 to 60 percent of cells across many organs. Many cell types maintained the corrected DNA sequence for at least six months—in fact, the most vulnerable cells in large arteries actually showed an almost 100 percent correction at 6 months, apparently because the corrected cells had compensated for the uncorrected cells that had died out. What’s more, the lifespan of the treated animals increased from seven to almost 18 months. In healthy mice, that’s approximately the beginning of old age.
This is the second notable advance in therapeutics for progeria in just three months. Last November, based on preclinical work from my lab and clinical trials conducted by the Progeria Research Foundation in Boston, the Food and Drug Administration (FDA) approved the first treatment for the condition. It is a drug called Zokinvy, and works by reducing the accumulation of progerin [5]. With long-term treatment, the drug is capable of extending the life of kids with progeria by 2.5 years and sometimes more. But it is not a cure.
We are hopeful this gene editing work might eventually lead to a cure for progeria. But mice certainly aren’t humans, and there are still important steps that need to be completed before such a gene-editing treatment could be tried safely in people. In the meantime, base editors and other gene editing approaches keep getting better—with potential application to thousands of genetic diseases where we know the exact gene misspelling. As we look ahead to 2021, the dream envisioned all those years ago about fixing the tiny DNA typo responsible for progeria is now within our grasp and getting closer to landing in the “can do” category.
References:
[1] In vivo base editing rescues Hutchinson-Gilford Progeria Syndrome in mice. Koblan LW et al. Nature. 2021 Jan 6.
[2] Base editor repairs mutation found in the premature-ageing syndrome progeria. Vermeij WP, Hoeijmakers JHJ. Nature. 6 Jan 2021.
[3] Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Nature. 2016 May 19;533(7603):420-424.
[4] Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR. Nature. 2017 Nov 23;551(7681):464-471.
[5] FDA approves first treatment for Hutchinson-Gilford progeria syndrome and some progeroid laminopathies. Food and Drug Administration. 2020 Nov 20.
Links:
Progeria (Genetic and Rare Diseases Information Center/NIH)
What are Genome Editing and CRISPR-Cas9? (National Library of Medicine/NIH)
Somatic Cell Genome Editing Program (Common Fund/NIH)
David R. Liu (Harvard University, Cambridge, MA)
Collins Group (National Human Genome Research Institute/NIH)
Jonathan Brown (Vanderbilt University Medical Center, Nashville, TN)
NIH Support: National Human Genome Research Institute; National Center for Advancing Translational Sciences; National Institute of Biomedical Imaging and Bioengineering; National Institute of Allergy and Infectious Diseases; National Institute of General Medical Sciences; Common Fund
Experts Conclude Heritable Human Genome Editing Not Ready for Clinical Applications
Posted on by Dr. Francis Collins

We stand at a critical juncture in the history of science. CRISPR and other innovative genome editing systems have given researchers the ability to make very precise changes in the sequence, or spelling, of the human DNA instruction book. If these tools are used to make non-heritable edits in only relevant tissues, they hold enormous potential to treat or even cure a wide range of devastating disorders, such as sickle cell disease, inherited neurologic conditions, and muscular dystrophy. But profound safety, ethical, and philosophical concerns surround the use of such technologies to make heritable changes in the human genome—changes that can be passed on to offspring and have consequences for future generations of humankind.
Such concerns are not hypothetical. Two years ago, a researcher in China took it upon himself to cross this ethical red line and conduct heritable genome editing experiments in human embryos with the aim of protecting the resulting babies against HIV infection. The medical justification was indefensible, the safety issues were inadequately considered, and the consent process was woefully inadequate. In response to this epic scientific calamity, NIH supported a call by prominent scientists for an international moratorium on human heritable, or germline, genome editing for clinical purposes.
Following on the heels of this unprecedented ethical breach, the U.S. National Academy of Sciences, U.S. National Academy of Medicine, and the U.K. Royal Society convened an international commission, sponsored by NIH, to conduct a comprehensive review of the clinical use of human germline genome editing. The 18-member panel, which represented 10 nations and four continents, included experts in genome editing technology; human genetics and genomics; psychology; reproductive, pediatric, and adult medicine; regulatory science; bioethics; and international law. Earlier this month, this commission issued its consensus study report, entitled Heritable Human Genome Editing [1].
The commission was designed to bring together thought leaders around the globe to engage in serious discussions about this highly controversial use of genome-editing technology. Among the concerns expressed by many of us was that if heritable genome editing were allowed to proceed without careful deliberation, the enormous potential of non-heritable genome editing for prevention and treatment of disease could become overshadowed by justifiable public outrage, fear, and disgust.
I’m gratified to say that in its new report, the expert panel closely examined the scientific and ethical issues, and concluded that heritable human genome editing is too technologically unreliable and unsafe to risk testing it for any clinical application in humans at the present time. The report cited the potential for unintended off-target DNA edits, which could have harmful health effects, such as cancer, later in life. Also noted was the risk of producing so-called mosaic embryos, in which the edits occur in only a subset of an embryo’s cells. This would make it very difficult for researchers to predict the clinical effects of heritable genome editing in human beings.
Among the many questions that the panel was asked to consider was: should society ever decide that heritable gene editing might be acceptable, what would be a viable framework for scientists, clinicians, and regulatory authorities to assess the potential clinical applications?
In response to that question, the experts replied: heritable gene editing, if ever permitted, should be limited initially to serious diseases that result from the mutation of one or both copies of a single gene. The first uses of these technologies should proceed incrementally and with extreme caution. Their potential medical benefits and harms should also be carefully evaluated before proceeding.
The commission went on to stress that before such an option could be on the table, all other viable reproductive possibilities to produce an embryo without a disease-causing alteration must be exhausted. That would essentially limit heritable gene editing to the exceedingly rare instance in which both parents have two copies of a recessive, disease-causing gene variant. Or another quite rare instance in which one parent has two copies of an altered gene for a dominant genetic disorder, such as Huntington’s disease.
Recognizing how unusual both scenarios would be, the commission held out the possibility that some would-be parents with less serious conditions might qualify if 25 percent or less of their embryos are free of the disease-causing gene variant. A possible example is familial hypercholesterolemia (FH), in which people carrying a mutation in the LDL receptor gene have unusually high levels of cholesterol in their blood. If both members of a couple are affected, only 25 percent of their biological children would be unaffected. FH can lead to early heart disease and death, but drug treatment is available and improving all the time, which makes this a less compelling example. Also, the commission again indicated that such individuals would need to have already traveled down all other possible reproductive avenues before considering heritable gene editing.
A thorny ethical question that was only briefly addressed in the commission’s report is the overall value to be attached to a couple’s desire to have a biological child. That desire is certainly understandable, although other options, such an adoption or in vitro fertilization with donor sperm, are available. This seems like a classic example of the tension between individual desires and societal concerns. Is the drive for a biological child in very high-risk situations such a compelling circumstance that it justifies asking society to start down a path towards modifying human germline DNA?
The commission recommended establishing an international scientific advisory board to monitor the rapidly evolving state of genome editing technologies. The board would serve as an access point for scientists, legislators, and the public to access credible information to weigh the latest progress against the concerns associated with clinical use of heritable human genome editing.
The National Academies/Royal Society report has been sent along to the World Health Organization (WHO), where it will serve as a resource for its expert advisory committee on human genome editing. The WHO committee is currently developing recommendations for appropriate governance mechanisms for both heritable and non-heritable human genome editing research and their clinical uses. That panel could issue its guidance later this year, which is sure to continue this very important conversation.
Reference:
[1] Heritable Human Genome Editing, Report Summary, National Academy of Sciences, September 2020.
Links:
“Heritable Genome Editing Not Yet Ready to Be Tried Safely and Effectively in Humans,” National Academies of Sciences, Engineering, and Medicine news release, Sep. 3, 2020.
International Commission on the Clinical Use of Human Germline Genome Editing (National Academies of Sciences, Engineering, and Medicine/Washington, D.C.)
Video: Report Release Webinar , International Commission on the Clinical Use of Human Germline Genome Editing (National Academies of Sciences, Engineering, and Medicine)
National Academy of Sciences (Washington, D.C.)
National Academy of Medicine (Washington, D.C.)
The Royal Society (London)
Study in Africa Yields New Diabetes Gene
Posted on by Dr. Francis Collins
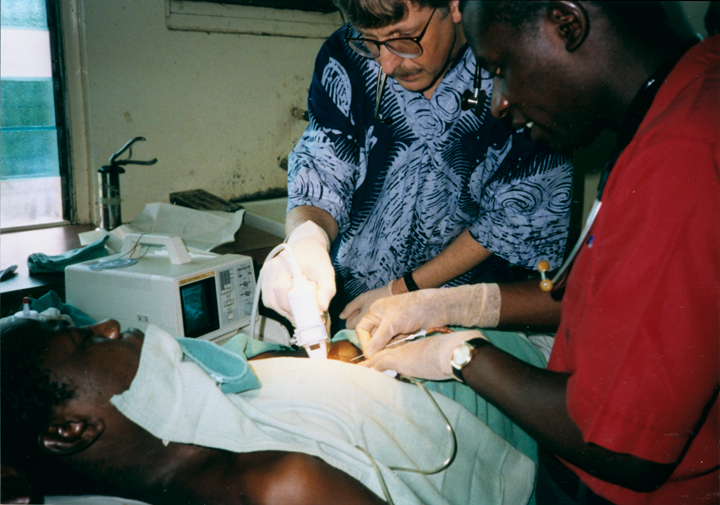
When I volunteered to serve as a physician at a hospital in rural Nigeria more than 25 years ago, I expected to treat a lot of folks with infectious diseases, such as malaria and tuberculosis. And that certainly happened. What I didn’t expect was how many people needed care for type 2 diabetes (T2D) and the health problems it causes. Surprisingly, these individuals were generally not overweight, and the course of their illness seemed different than in the West.
The experience inspired me to join with other colleagues at Howard University, Washington, DC, to help found the Africa America Diabetes Mellitus (AADM) study. It aims to uncover genomic risk factors for T2D in Africa and, using that information, improve understanding of the condition around the world.
So, I’m pleased to report that, using genomic data from more than 5,000 volunteers, our AADM team recently discovered a new gene, called ZRANB3, that harbors a variant associated with T2D in sub-Saharan Africa [1]. Using sophisticated laboratory models, the team showed that a malfunctioning ZRANB3 gene impairs insulin production to control glucose levels in the bloodstream.
Since my first trip to Nigeria, the number of people with T2D has continued to rise. It’s now estimated that about 8 to 10 percent of Nigerians have some form of diabetes [2]. In Africa, diabetes affects more than 7 percent of the population, more than twice the incidence in 1980 [3].
The causes of T2D involve a complex interplay of genetic, environmental, and lifestyle factors. I was particularly interested in finding out whether the genetic factors for T2D might be different in sub-Saharan Africa than in the West. But at the time, there was a dearth of genomic information about T2D in Africa, the cradle of humanity. To understand complex diseases like T2D fully, we need all peoples and continents represented in the research.
To begin to fill this research gap, the AADM team got underway and hasn’t looked back. In the latest study, led by Charles Rotimi at NIH’s National Human Genome Research Institute, in partnership with multiple African diabetes experts, the AADM team enlisted 5,231 volunteers from Nigeria, Ghana, and Kenya. About half of the study’s participants had T2D and half did not.
As reported in Nature Communications, their genome-wide search for T2D gene variants turned up three interesting finds. Two were in genes previously linked to T2D risk in other human populations. The third involved a gene that codes for ZRANB3, an enzyme associated with DNA replication and repair that had never been reported in association with T2D.
To understand how ZRANB3 might influence a person’s risk for developing T2D, the researchers turned to zebrafish (Danio rerio), an excellent vertebrate model for its rapid development. The researchers found that the ZRANB3 gene is active in insulin-producing beta cells of the pancreas. That was important to know because people with T2D frequently have reduced numbers of beta cells, which compromises their ability to produce enough insulin.
The team next used CRISPR/Cas9 gene-editing tools either to “knock out” or reduce the expression of ZRANB3 in young zebrafish. In both cases, it led to increased loss of beta cells.
Additional study in the beta cells of mice provided more details. While normal beta cells released insulin in response to high levels of glucose, those with suppressed ZRANB3 activity couldn’t. Together, the findings show that ZRANB3 is important for beta cells to survive and function normally. It stands to reason, then, that people with a lower functioning variant of ZRANB3 would be more susceptible to T2D.
In many cases, T2D can be managed with some combination of diet, exercise, and oral medications. But some people require insulin to manage the disease. The new findings suggest, particularly for people of African ancestry, that the variant of the ZRANB3 gene that one inherits might help to explain those differences. People carrying particular variants of this gene also may benefit from beginning insulin treatment earlier, before their beta cells have been depleted.
So why wasn’t ZRANB3 discovered in the many studies on T2D carried out in the United States, Europe, and Asia? It turns out that the variant that predisposes Africans to this disease is extremely rare in these other populations. Only by studying Africans could this insight be uncovered.
More than 20 years ago, I helped to start the AADM project to learn more about the genetic factors driving T2D in sub-Saharan Africa. Other dedicated AADM leaders have continued to build the research project, taking advantage of new technologies as they came along. It’s profoundly gratifying that this project has uncovered such an impressive new lead, revealing important aspects of human biology that otherwise would have been missed. The AADM team continues to enroll volunteers, and the coming years should bring even more discoveries about the genetic factors that contribute to T2D.
References:
[1] ZRANB3 is an African-specific type 2 diabetes locus associated with beta-cell mass and insulin response. Adeyemo AA, Zaghloul NA, Chen G, Doumatey AP, Leitch CC, Hostelley TL, Nesmith JE, Zhou J, Bentley AR, Shriner D, Fasanmade O, Okafor G, Eghan B Jr, Agyenim-Boateng K, Chandrasekharappa S, Adeleye J, Balogun W, Owusu S, Amoah A, Acheampong J, Johnson T, Oli J, Adebamowo C; South Africa Zulu Type 2 Diabetes Case-Control Study, Collins F, Dunston G, Rotimi CN. Nat Commun. 2019 Jul 19;10(1):3195.
[2] Diabetes mellitus in Nigeria: The past, present and future. Ogbera AO, Ekpebegh C. World J Diabetes. 2014 Dec 15;5(6):905-911.
[3] Global report on diabetes. Geneva: World Health Organization, 2016. World Health Organization.
Links:
Diabetes (National Institute of Diabetes ad Digestive and Kidney Diseases/NIH)
Diabetes and African Americans (Department of Health and Human Services)
Why Use Zebrafish to Study Human Diseases (Intramural Research Program/NIH)
Charles Rotimi (National Human Genome Research Institute/NIH)
NIH Support: National Human Genome Research Institute; National Institute of Diabetes and Digestive and Kidney Diseases; National Institute on Minority Health and Health Disparities
Study Shows Genes Unique to Humans Tied to Bigger Brains
Posted on by Dr. Francis Collins
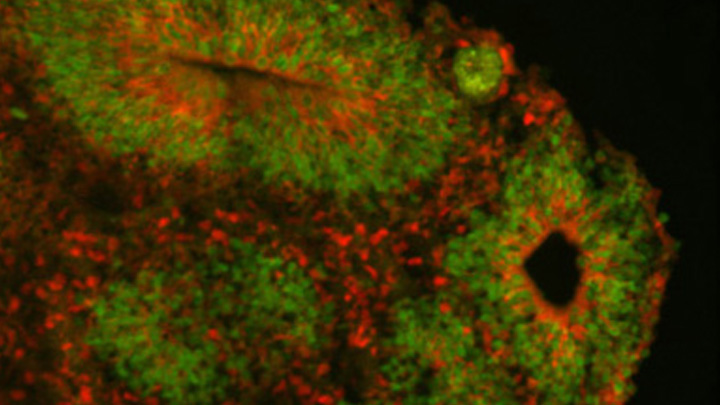
Caption: Cortical organoid, showing radial glial stem cells (green) and cortical neurons (red).
Credit: Sofie Salama, University of California, Santa Cruz
In seeking the biological answer to the question of what it means to be human, the brain’s cerebral cortex is a good place to start. This densely folded, outer layer of grey matter, which is vastly larger in Homo sapiens than in other primates, plays an essential role in human consciousness, language, and reasoning.
Now, an NIH-funded team has pinpointed a key set of genes—found only in humans—that may help explain why our species possesses such a large cerebral cortex. Experimental evidence shows these genes prolong the development of stem cells that generate neurons in the cerebral cortex, which in turn enables the human brain to produce more mature cortical neurons and, thus, build a bigger cerebral cortex than our fellow primates.
That sounds like a great advantage for humans! But there’s a downside. Researchers found the same genomic changes that facilitated the expansion of the human cortex may also render our species more susceptible to certain rare neurodevelopmental disorders.
Creative Minds: Building a CRISPR Gene Drive Against Malaria
Posted on by Dr. Francis Collins

Valentino Gantz/Credit: Erik Jepsen
Researchers have used Drosophila melanogaster, the common fruit fly that sometimes hovers around kitchens, to make seminal discoveries involving genetics, the nervous system, and behavior, just to name a few. Could a new life-saving approach to prevent malaria be next? Valentino Gantz, a researcher at the University of California, San Diego, is on a path to answer that question.
Gantz has received a 2016 NIH Director’s Early Independence Award to use Drosophila to hone a new bioengineered tool that acts as a so-called “gene drive,” which spreads a new genetically encoded trait through a population much faster than would otherwise be possible. The lessons learned while working with flies will ultimately be applied to developing a more foolproof system for use in mosquitoes with the hope of stopping the transmission of malaria and potentially other serious mosquito-borne diseases.
Gene Drive Research Takes Aim at Malaria
Posted on by Dr. Francis Collins
 Malaria has afflicted humans for millennia. Even today, the mosquito-borne, parasitic disease claims more than a half-million lives annually [1]. Now, in a study that has raised both hope and concern, researchers have taken aim at this ancient scourge by using one of modern science’s most powerful new technologies—the CRISPR/Cas9 gene-editing tool—to turn mosquitoes from dangerous malaria vectors into allies against infection [2].
Malaria has afflicted humans for millennia. Even today, the mosquito-borne, parasitic disease claims more than a half-million lives annually [1]. Now, in a study that has raised both hope and concern, researchers have taken aim at this ancient scourge by using one of modern science’s most powerful new technologies—the CRISPR/Cas9 gene-editing tool—to turn mosquitoes from dangerous malaria vectors into allies against infection [2].
The secret behind this new strategy is the “gene drive,” which involves engineering an organism’s genome in a way that intentionally spreads, or drives, a trait through its population much faster than is possible by normal Mendelian inheritance. The concept of gene drive has been around since the late 1960s [3]; but until the recent arrival of highly precise gene editing tools like CRISPR/Cas9, the approach was largely theoretical. In the new work, researchers inserted into a precise location in the mosquito chromosome, a recombinant DNA segment designed to block transmission of malaria parasites. Importantly, this segment also contained a gene drive designed to ensure the trait was inherited with extreme efficiency. And efficient it was! When the gene-drive engineered mosquitoes were mated with normal mosquitoes in the lab, they passed on the malaria-blocking trait to 99.5 percent of their offspring (as opposed to 50 percent for Mendelian inheritance).
Flipping a Genetic Switch on Obesity?
Posted on by Dr. Francis Collins
 When weight loss is the goal, the equation seems simple enough: consume fewer calories and burn more of them exercising. But for some people, losing and keeping off the weight is much more difficult for reasons that can include a genetic component. While there are rare genetic causes of extreme obesity, the strongest common genetic contributor discovered so far is a variant found in an intron of the FTO gene. Variations in this untranslated region of the gene have been tied to differences in body mass and a risk of obesity [1]. For the one in six people of European descent born with two copies of the risk variant, the consequence is carrying around an average of an extra 7 pounds [2].
When weight loss is the goal, the equation seems simple enough: consume fewer calories and burn more of them exercising. But for some people, losing and keeping off the weight is much more difficult for reasons that can include a genetic component. While there are rare genetic causes of extreme obesity, the strongest common genetic contributor discovered so far is a variant found in an intron of the FTO gene. Variations in this untranslated region of the gene have been tied to differences in body mass and a risk of obesity [1]. For the one in six people of European descent born with two copies of the risk variant, the consequence is carrying around an average of an extra 7 pounds [2].
Now, NIH-funded researchers reporting in The New England Journal of Medicine [3] have figured out how this gene influences body weight. The answer is not, as many had suspected, in regions of the brain that control appetite, but in the progenitor cells that produce white and beige fat. The researchers found that the risk variant is part of a larger genetic circuit that determines whether our bodies burn or store fat. This discovery may yield new approaches to intervene in obesity with treatments designed to change the way fat cells handle calories.
Popular Genome Editing Tool Gets Its Close-Up
Posted on by Dr. Francis Collins
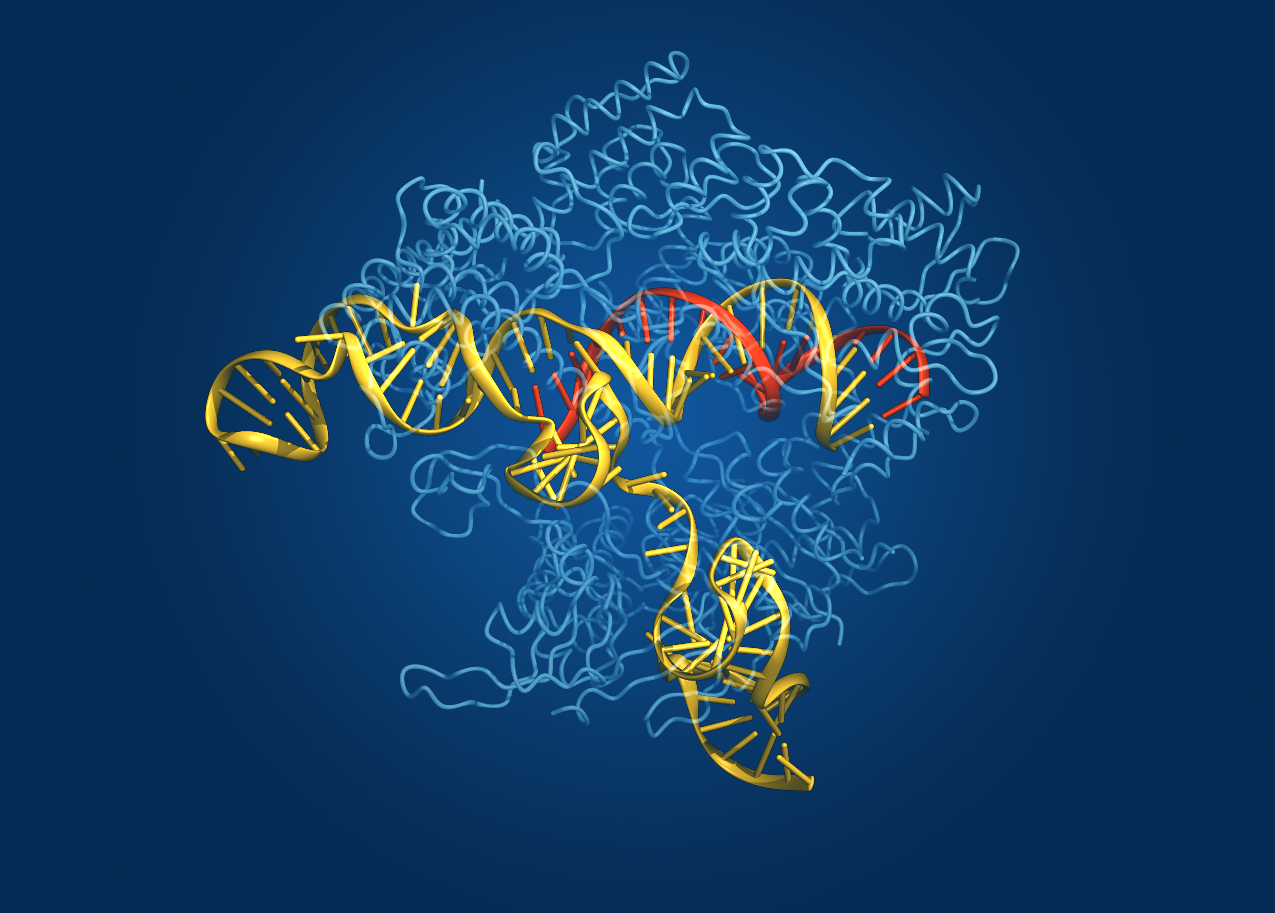
Caption: Crystal structure of the Cas9 gene-editing enzyme (light blue) in complex with an RNA guide (red) and its target DNA (yellow).
Credit: Bang Wong, Broad Institute of Harvard and MIT, Cambridge, MA
Exactly one hundred years ago, Max von Laue won the Nobel Prize in Physics for discovering that when a crystal is bombarded with X-rays, the beams bounce off the electrons surrounding the nucleus of each atom and scatter, interfering with each other (like ripples in a pond) and creating a unique pattern. These diffraction patterns could be used to decipher the arrangement of atoms in the crystal. Since then, X-ray crystallography has been used to chart a vast number of biological structures, including those of DNA, proteins, and even whole viruses.
Now, NIH-funded researchers at the Broad Institute of MIT and Harvard (Cambridge, MA) have teamed up with researchers at the University of Tokyo (Japan) to use crystallography to generate a high-definition map of an innovative tool for editing genomes. Their image reveals the structure of Cas9—an enzyme with an amazing ability to slice DNA with exquisite precision—in complex with a molecule of RNA that is guiding it to a targeted region of DNA [1].
The Cas9 enzyme was originally discovered in bacteria. It’s a key part of an ancient microbial immune system, called CRISPR-Cas (Clustered Regularly Interspaced Short Palindromic Repeats-Cas), that researchers recently discovered could be put to use as a tool for precisely altering DNA. This extraordinary system has been used to knock out genes in cells from bacteria, mice, and humans, and even to engineer monkeys with specific mutations that could serve as more accurate models of human disease.
Still, there’s room for improvement. Because Cas9 is rather large, Broad researcher Feng Zhang (a recipient of both the NIH Director’s Transformative Research Award and an NIH Director’s Pioneer Award) wants to trim the enzyme a bit so it could be packaged into viruses for new applications. Armed with the new crystal structure, Zhang’s team can now determine which regions of the enzyme are essential for editing DNA and which parts might be dispensable.
Another issue with Cas9 is that it occasionally makes errors, cutting the wrong region of DNA. Zhang thinks the new structural schematic of Cas9, along with the guide molecule RNA and target DNA, might point to ways in which the enzyme can be optimized to reduce the chance of errors.
Zhang’s team isn’t the only one interested in tweaking Cas9 to improve its engineering potential. A group led by Jennifer Doudna and Eva Nogales, both of the University of California, Berkeley, also recently used X-ray crystallography to generate images of two different versions of Cas9: one from Streptococcus pyogenes and the other from Actinomyces naeslundii [2]. By the way, the Foundation for the NIH recently named Doudna as the winner of its 2014 Lurie Prize in the Biomedical Sciences. The NIH grantee received the award for her pioneering role in the 2012 discovery of the CRISPR gene-editing technique.
Thanks to all of these new crystal structures, the scientific community is a step closer to realizing the full potential of Cas9/CRISPR technology to advance our understanding of disease and accelerate development of treatments and cures. So, here’s to our old friend crystallography and all of the exciting ways in which it will continue to expand our scientific horizons for years to come!
References:
[1] Crystal Structure of Cas9 in Complex with Guide RNA and Target DNA. Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O. Cell. 2014 Feb 12. pii: S0092-8674(14)00156-1.
[2] Structures of Cas9 Endonucleases Reveal RNA-Mediated Conformational Activation. Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E, Doudna JA. Science. 2014 Feb 6
Links:
Zhang Lab, Broad Institute of Harvard and MIT, Cambridge, MA
Genome Engineering Resource Center maintained by the Zhang Lab
Nureki Lab, Department of Biophysics and Biochemistry, The University of Tokyo
Doudna Lab, University of California, Berkeley, CA
Foundation for the NIH to Award Lurie Prize in Biomedical Sciences to Jennifer Doudna from UC Berkeley, 25 February 2014
The Nogales Lab, University of California, Berkeley, CA
NIH Director’s Pioneer Award. (NIH Common Fund)
NIH Director’s Transformative Research Award. (NIH Common Fund)
NIH support: Office of the Director (Common Fund); National Institute of Mental Health; National Institute of General Medical Sciences
Copy-editing the Genome: Extreme Personalized Medicine?
Posted on by Dr. Francis Collins
COOL TOOL. See how the TALE protein (rainbow colored) recognizes the target DNA site and wraps around the double helix. When this TALE protein is fused to a nuclease (the scissors), creating a TALEN, the hybrid protein will clip the DNA at the target site. Credit: Jeffry D. Sander, Massachusetts General Hospital |
If I made a spelling mistake in this blog, and you were my copy editor, you’d want to fix it quickly. You’d delete the wrong letter and insert the correct one. Well, DNA is a language too, with just four letters in its alphabet; and disease can occur with just one letter out of place if it’s in a vulnerable position (think sickle cell anemia or the premature aging disease, progeria). Wouldn’t it be great for tomorrow’s physicians to be able to do what the copy editor does? That is, if they could fix a genetic mutation quickly and efficiently, without messing up the rest of the text?
Previous Page Next Page